
Environmentally benign diastereoselective synthesis of granatane and tropane aldol derivatives†
Abstract
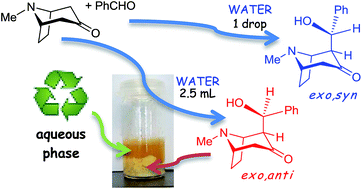
Direct aldol reactions of tropinone and granatanone (pseudopelletierine) with aromatic aldehydes were promoted by the presence of water. The anti–syn-diastereoselectivity depended on the amount of water used or on the possibility of product precipitation from the reaction mixture. Some of the reactions showed excellent atom economy, a low E factor, and high diastereoselectivity (up to 98%). In several cases ‘seeding’ with the anti isomer, used to induce the deposition of solid products, improved the conversion (up to 1.8 times) and the anti–syn ratio (up to 98 : 2). The applicability of spontaneous direct aldol reactions in the presence of water was also extended to N-alkyl nortropanones or norgranatanones using an aqueous–organic medium. However, under these conditions only the exo,syn isomers of the N-substituted aldols were obtained. The syn-selectivity for the tropane- and granatane-related aldols is specific for water-promoted reactions and results from thermodynamic control.
 Please wait while we load your content...
Please wait while we load your content...

