NHC catalysed direct addition of HMF to diazo compounds: synthesis of acyl hydrazones with antitumor activity†
João P. M. Antónioa,
Raquel F. M. Fradea,
Fábio M. F. Santosa,
Jaime A. S. Coelhoa,
Carlos A. M. Afonsoa,
Pedro M. P. Gois*a and
Alexandre F. Trindade*ab
aInstituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia, Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal. E-mail: alexandretrindade@ff.ul.pt; pedrogois@ff.ul.pt; Fax: +351 217 946 470
bCQFM, Centro de Química-Física Molecular, IN-Institute of Nanosciences and Nanotechnology, Instituto Superior Técnico, 1049-001 Lisboa, Portugal. Fax: +351 218 464 455/7
First published on 23rd June 2014
Abstract
NHC umpolung catalysis between 5-hydroxymethyl furfural (HMF) derivatives and diazo compounds overcomes the usual multistep synthesis of acylhydrazones and gives direct access to an unexplored family of HMF-based acylhydrazones displaying promising anti-tumor activity. A preliminary screening of hydroxymethyl's protection groups allowed the identification of the tert-butyldimethylsilyl group as being essential for the desired biological activity. Compound 25 was found to be very active against MCF-7 breast cell line (with an IC50s of 3.60 μM) and while exerting a much lower toxicity in differentiated CaCo-2 monolayer.
Introduction
Hydrazones are an important scaffold for medicinal chemistry, present in a wide range of molecules with antimicrobial, anti-inflammatory, antiviral and antitumor properties.1 Among these, acylhydrazones are particular interesting because they often display high cytotoxic activity.2–11 For instance, PAC-1 is a potent cytotoxic acylhydrazone that was recently approved for clinical trials (Scheme 1). This molecule is thought to induce apoptosis via the activation of procaspase-3 interfering in the zinc-mediated inhibition pathway.12,13 Recognizing the importance of these molecules to effectively chelate metal ions, the furan moiety was included in the structure of several acylhydrazones to improve their antitumor activity. This strategy lead to the synthesis of several 5-aryl-N-furanoyl hydrazones with promising cytotoxic activities against human promyelocytic leukemic cells (15 μM (IC50) for HL-60, Scheme 1).14 Based on this rational we conceived that further manipulation of this scaffold to include additional chelating sub units, could render N-furanoyl hydrazones with improved biological activity (Scheme 1). | ||
| Scheme 1 PAC-1 structure; N-acyl hydrazones with antitumoral activity; conventional versus the application of NHC organocatalysis in the synthesis of novel HMF-based N-acyl hydrazones with potential antitumor activity. | ||
5-Hydroxymethyl furfural (HMF) is a very important synthetic motif which has recently gathered considerable attention for its unique structure and biorenewable origins.15–19 As shown in Scheme 1, HMF exhibits a substituted furan ring ideally suited to build structurally diverse N-furanoyl hydrazones. Despite this, HMF synthetic manipulation is often troublesome due to a limited chemical and thermal stability.17,20 For instance, in acidic conditions, HMF suffers hydrolysis and formic acid extrusion to yield levulinic acid, while in basic conditions, it readily affords 5-hydroxymethyl furanoic acid and 2,5-dihydroxymethyl furan via a Cannizzaro reaction.21,22 Therefore, traditional methods to prepare hydrazones became very problematic when using HMF and require a multistep sequence comprising the protection of the alcohol, oxidation of the aldehyde, activation of the carboxylic acid and deprotection of the alcohol (Scheme 1). Hence we conceive that an alternative strategy to circumvent this problem would be the direct addition of the aldehyde to a diazo compound catalysed by N-heterocyclic carbenes (NHC).23 Based on our recent experience on the use of NHCs as organocatalysts23–25 and in the synthesis and manipulation of HMF,15,16,21 we initiated a study to establish if NHCs could be effectively used to umpolung the reactivity of HMF's aldehyde and to catalyse the direct synthesis of N-furanoyl hydrazones with improved cytotoxic activity against model cancer cell lines.
Results and discussion
We initiated this study by testing the NHC catalysed direct addition of HMF 1 to diazo compound 2. Based on our previous study on the addition of aldehydes to diazo compounds, we identified NHCs generated from imidazolidinium 4, imidazolium 5 and triazolium 6 as the most promising catalysts for this transformation (Scheme 2).23 Therefore HMF 1 was slowly added to a pre-stirred solution of 20 mol% of each of the NHC precursors, 40 mol% of DBU and diazo compound 2. Differently from our previous study, NHCs generated in situ from 5 and 6 performed poorly when using HMF as the starting material, affording the desired N-acyl hydrazone in only 11 and <5% yield respectively. Differently and to our delight, when using imidazolidinium 4 as the NHC precursor, the desired compound 3 was obtained in 70% isolated yield after 16 hours at room temperature (Table 1, entry 1).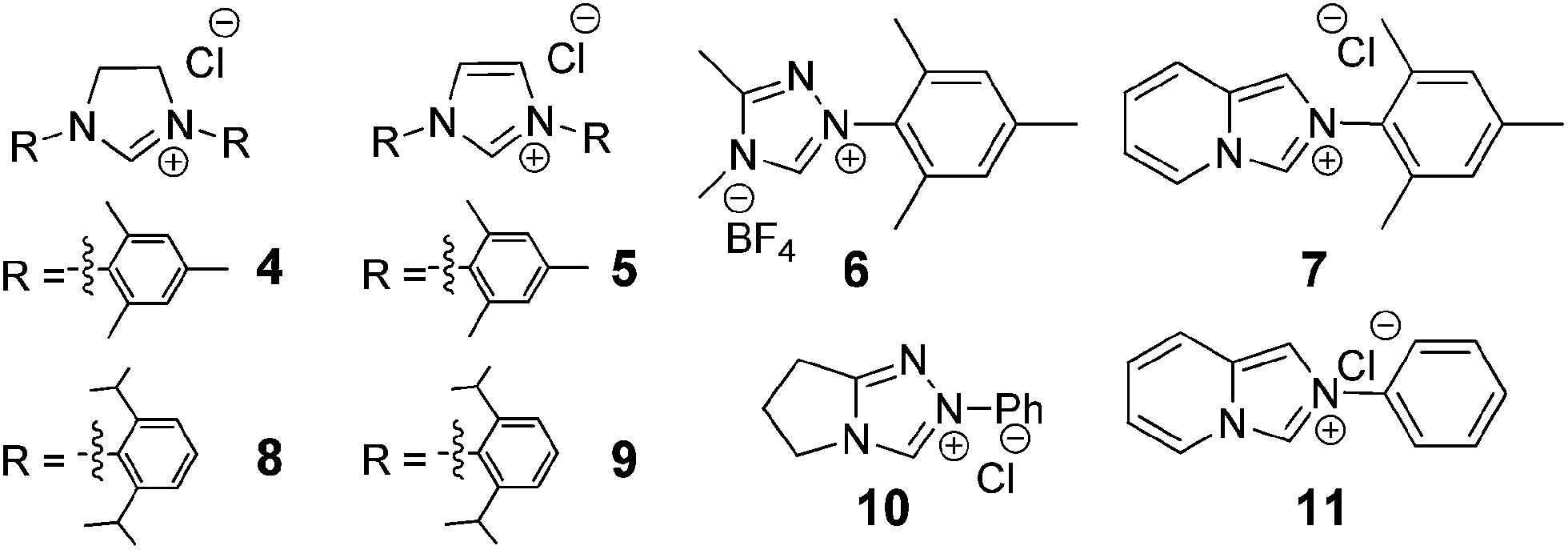 | ||
| Scheme 2 NHC precursors employed in this work. | ||
|
||||
|---|---|---|---|---|
| Entry | NHC precursor | Base | Solvent | Yield (%) |
| a HMF 1 (1.5 eq.), diazo 2 (1 eq.), NHC precursor (20 mol%), base (40 mol%), aldehyde slow addition (0.2 mL solvent in 30 minutes).b Identical to (a) except base (20 mol%).c Identical to (a) except NHC precursor (10 mol%), base (20 mol%).d Identical to (a) except aldehyde slow addition (0.5 mL solvent in 2 hours); n.r. – no reaction; DBU – 1,8-diazabicyclo[5.4.0]undec-7-ene; TEA – triethyl amine DIPEA – diisopropyl ethyl amine. | ||||
| 1a | 4 | DBU | CH2Cl2 | 70 |
| 2a | 5 | DBU | CH2Cl2 | 11 |
| 3a | 6 | DBU | CH2Cl2 | 6 |
| 4a | 7 | DBU | CH2Cl2 | <5 |
| 5a | 8 | DBU | CH2Cl2 | 8 |
| 6a | 9 | DBU | CH2Cl2 | 5 |
| 7a | 10 | DBU | CH2Cl2 | <5 |
| 8a | 11 | DBU | CH2Cl2 | <5 |
| 9a | 4 | NaH | CH2Cl2 | <5 |
| 10a | 4 | NaOtBu | CH2Cl2 | <5 |
| 11a | 4 | TEA | CH2Cl2 | <5 |
| 12a | 4 | DIPEA | CH2Cl2 | <5 |
| 13a | 4 | DBU | THF | 41 |
| 14a | 4 | DBU | CH3CN | 40 |
| 15a | 4 | DBU | MeOH | <5 |
| 16a | 4 | DBU | Toluene | n.r. |
| 17b | 4 | DBU (20) | CH2Cl2 | 49 |
| 18c | 4 (10) | DBU (20) | CH2Cl2 | 38 |
| 19d | 4 | DBU | CH2Cl2 | 80 |

This compound was shown to be a unique organocatalyst for this amidation reaction as all other NHCs tested afforded only traces of compound 3 (Table 1, entries 4–8). Once established the NHC generated in situ from imidazolidinium 4 as the most effective organocatalyst precursor to perform the addition of HMF to diazo compound 2, we addressed the optimization of the reaction conditions. As shown in Table 1, when using bases like NaH, NaOtBu, triethylamine and diisopropylethyl amine (Table 1, entries 9–12) the yield of hydrazone 3 decreased considerably. The reaction was also less efficient when performed in solvents such as THF and acetonitrile (Table 1, entries 13–14) and was completely inhibited in methanol and toluene (Table 1, entries 15–16). Similarly reducing the amount of base or organocatalyst to 20 mol% and 10 mol% respectively resulted in lower yields of N-acyl hydrazone (Table 1, entry 17 and 18). Finally, by increasing the aldehyde addition time, the isolated yield of the compound 3 was improved to 80% (Table 1, entry 19). Interestingly, the analysis of the reaction crude mixture by HPLC (see ESI†) did not indicate the formation of the expected competitive Cannizzaro products (2,5-dihydroxymethyl furan or 5-hydroxymethyl furoic acid) neither the products resulting from the NHC catalysed self-condensation of HMF. After establishing the optimised reaction conditions and aiming at the synthesis of structurally different N-(5-hydroxymethyl furfuryl) hydrazones for biological evaluation, we tested the addition of HMF to several diazo compounds.
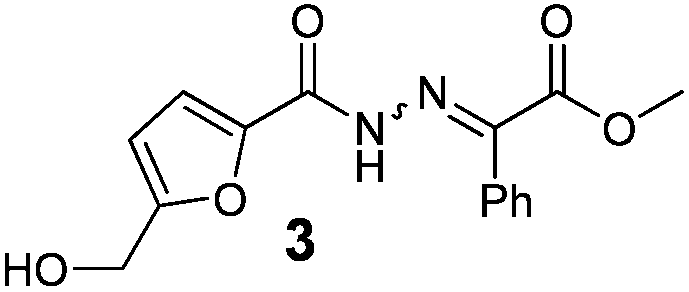
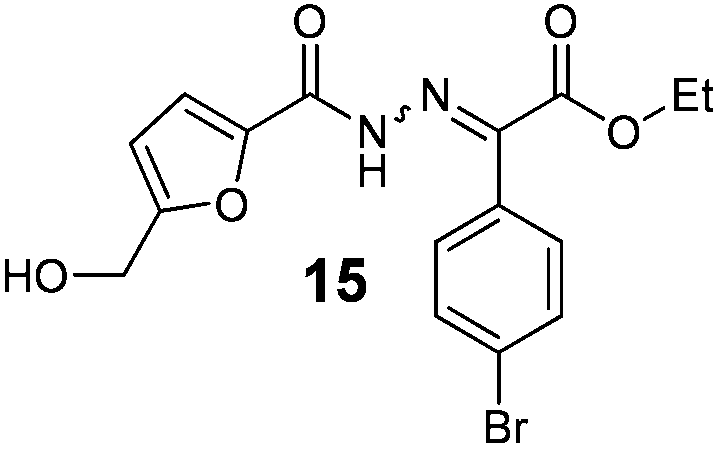
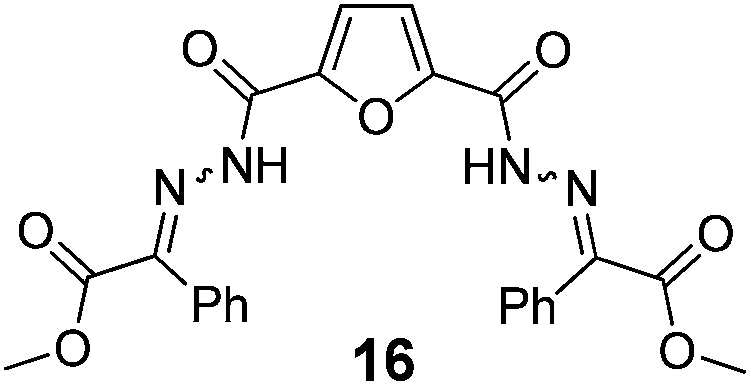
As shown in Table 2, using diazo compounds featuring different aromatic and ester substituents afforded the expected N-(5-hydroxymethyl furfuryl) hydrazones in moderate to good yields (42–80%) as a mixture of geometric isomers E/Z (for more details consult ESI†). Furan-2,5-dicarbaldehyde was also shown to be a viable substrate for this transformation, yielding the symmetrical N-acyl hydrazone 16 in 53% yield (Table 2, entry 6). Once achieved the synthesis of HMF derived N-acyl hydrazones 3, 12–16, we envisioned that further modification of the hydrazone structure could be obtained by modifying the hydroxyl group at the HMF. Therefore, we tested the direct addition of O-protected HMF derivatives to diazo compounds under the optimized reaction conditions. As shown in Scheme 3, the ester derivatives displayed lower reactivity than HMF itself delivering hydrazones 17–19 in up to 34% yield (Scheme 3, top). Very differently, both benzyl and TBDMS ethers afforded the expected products in 89 and 99% isolated yields respectively (Scheme 3). With compounds 3, 12–16, 20 and 21 in hands we performed the evaluation of their anti-proliferative properties against HT-29, MCF-7 and NCI-H460 cancer cell lines at concentrations within the range 0–15 μM (Table 3). Very disappointingly, N-(5-hydroxymethyl furfuryl) hydrazones 3, 12–16 didn't exhibit any relevant biological activity, similarly HMF benzyl ether derivative 20 was also unable to prevent the proliferation of any of the cancer cell lines at these concentrations. Differently, the HMF TBDMS ether 21 proved to be active against MCF-7 cancer cell line with an IC50 of 13.32 μM. These results indicate that the presence of the TBDMS group on the molecule structure is important for the biological activity probably due to an increase of the compound lipophilicity. This is in line with previous observations for other scaffolds and biological activities.26 Therefore, compounds 22–26 were readily prepared under our optimized reaction conditions and tested against the same cancer cell lines. As shown in Table 3, compounds 22 and 23 were shown to be inactive while compound 24 inhibit the proliferation of this cancer cell models in the range of 8.59–13.32 μM IC50s. Very gratifyingly, these results were further improved with compounds 25 and 26 that displayed IC50s in the interval of 3.60–6.37 and 3.50–7.29 μM, respectively. Furthermore compounds 25 and 26 also displayed the lowest toxicities towards differentiated CaCo-2 monolayer, a model that mimics human intestinal epithelium, as demonstrated by the highest IC50s (40.12 and 27.90 μM, respectively).

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:



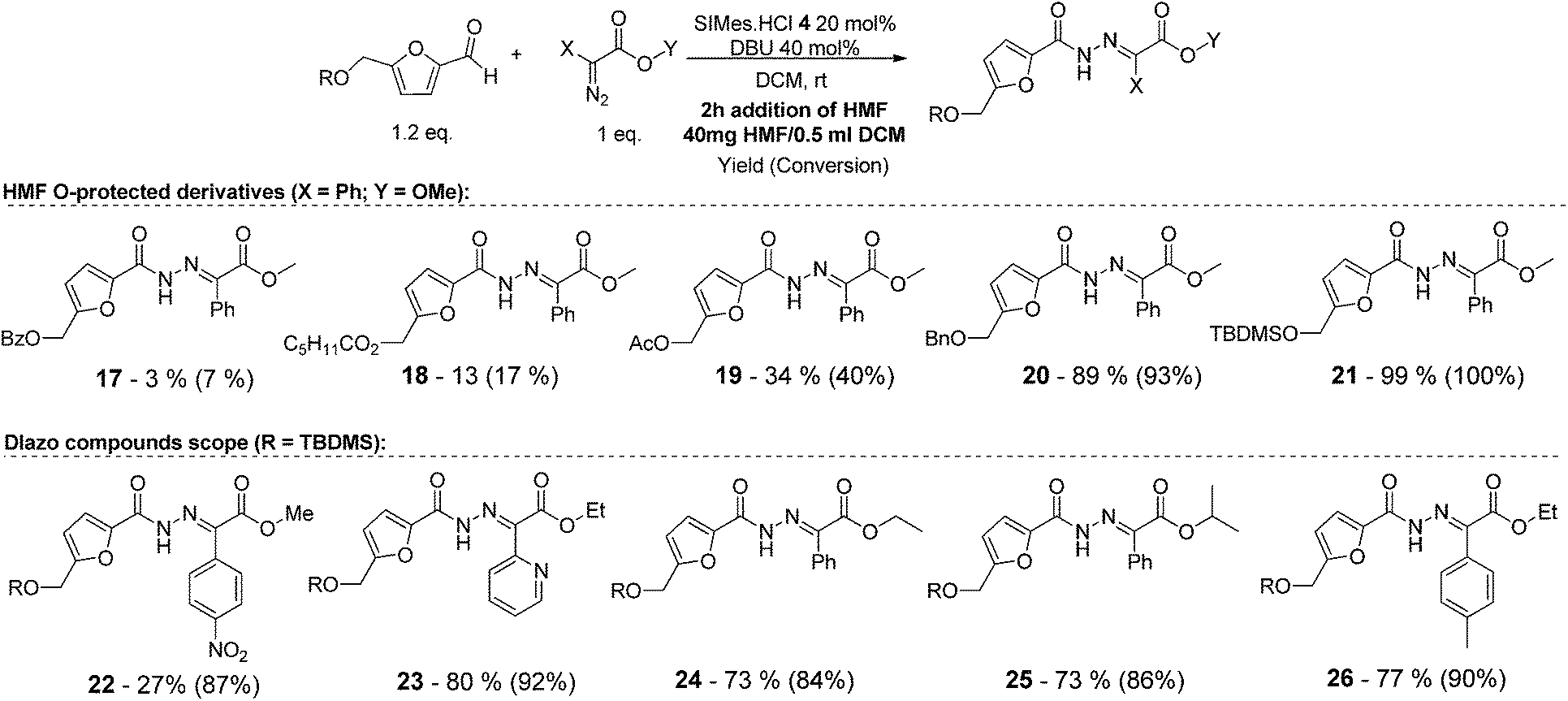 | ||
| Scheme 3 Substrate scope of the NHC-catalysed formation of N-acyl hydrazones from HMF O-derivatives. | ||
| IC50 μM | HT-29 | MCF-7 | NCI-H460 | CaCo-2 |
|---|---|---|---|---|
| 3 | >15 | >15 | >15 | — |
| 12 | >15 | >15 | >15 | — |
| 13 | >15 | >15 | >15 | — |
| 14 | >15 | >15 | >15 | — |
| 15 | >15 | >15 | >15 | — |
| 16 | >15 | >15 | >15 | — |
| 20 | >15 | >15 | >15 | — |
| 21 | >15 | 13.32 (1.44) | >15 | 19.14 |
| 22 | >15 | >15 | >15 | — |
| 23 | >15 | >15 | >15 | — |
| 24 | 8.59 (1.82) | 13.32 (1.17) | 10.71 (1.53) | 15.62 |
| 25 | 6.37 (6.30) | 3.60 (11.14) | 5.06 (6.73) | 40.12 |
| 26 | 7.29 (3.82) | 3.50 (7.97) | 6.95 (4.01) | 27.90 |
Conclusions
The HMF scaffold offers the possibility to access a diverse and unexplored family of acylhydrazones with potential antitumor activity. However, the utilization of HMF for the synthesis of higher value molecules such as fine chemicals or drugs is a challenging area of research due to its peculiar chemistry and thermal and/or chemical stability. This motivated the discovery of a new method to directly access acylhydrazones from the HMF scaffold overcoming the common multistep synthesis.Herein, it was demonstrated that NHCs are able to selectively catalyse the nucleophilic addition of HMF and its O-protected derivatives to aryl diazoacetates under mildly basic conditions at room temperature affording the desired acylhydrazones, while avoiding the usual degradation pathways of HMF. From our preliminary biological evaluation studies compounds 25 and 26 were identified as active cytotoxic agents (displayed IC50s in the interval of 3.60–6.37 and 3.50–7.29 μM, respectively) indicating that t-butyldimethylsilyl group is essential for the desired biological activity. Additionally compound 25 displayed an increased 10-fold selectivity towards differentiated CaCo-2 monolayer inducing an IC50 of 40.12 μM.
Experimental
Synthesis of HMF-based N-acylhydrazones
NHC precursor 4 (20 mol%) and DBU (40 mol%) were dissolved in freshly dry DCM (0.5 mL) inside a round bottom flask under argon. The mixture was stirred for 10 minutes at room temperature. Then a solution of diazo compound (0.24 mmol in 0.5 mL of DCM) was added. Immediately after, HMF (1.2 eq., 40 mg in 0.5 mL of DCM) (or protected HMF) was slowly added over two hours. The mixture was left reacting until most of diazo compound was consumed, usually 16 hours. The final product, N-acylhydrazone, was isolated by flash chromatography (eluent: hexane/ethyl acetate).Acknowledgements
We thank the Fundação para a Ciência e Tecnologia for financial support (SFRH/BPD/73932/2010, SFRH/BD/73971/2010, SFRH/BD/94779/2013, PTDC/QUI-QUI/119823/2010, PTDC/QUI-QUI/118315/2010, PEst-OE/SAU/UI4013/2011, SFRH/BPD/73822/2010).Notes and references
- S. Rollas and Ş. G. Küçükgüzel, Molecules, 2007, 12, 1910–1939 CrossRef CAS.
- D. W. Wolan, J. A. Zorn, D. C. Gray and J. A. Wells, Science, 2009, 326, 853–858 CrossRef CAS PubMed.
- P. Vicini, M. Incerti, I. A. Doytchinova, P. La Colla, B. Busonera and R. Loddo, Eur. J. Med. Chem., 2006, 41, 624–632 CrossRef CAS PubMed.
- L. Jin, J. Chen, B. Song, Z. Chen, S. Yang, Q. Li, D. Hu and R. Xu, Bioorg. Med. Chem. Lett., 2006, 16, 5036–5040 CrossRef CAS PubMed.
- H. Z. Zhang, J. Drewe, B. Tseng, S. Kasibhatla and S. X. Cai, Bioorg. Med. Chem., 2004, 12, 3649–3655 CrossRef CAS PubMed.
- C. Congiu and V. Onnis, Bioorg. Med. Chem., 2013, 21, 6592–6599 CrossRef CAS PubMed.
- B. Zhang, Y. Zhao, X. Zhai, L. Wang, J. Yang, Z. Tan and P. Gong, Chem. Pharm. Bull., 2012, 60, 1046–1054 CrossRef CAS PubMed.
- A. R. Germain, L. C. Carmody, B. Morgan, C. Fernandez, E. Forbeck, T. A. Lewis, P. P. Nag, A. Ting, L. VerPlank, Y. Feng, J. R. Perez, S. Dandapani, M. Palmer, E. S. Lander, P. B. Gupta, S. L. Schreiber and B. Munoz, Bioorg. Med. Chem. Lett., 2012, 22, 3571–3574 CrossRef CAS PubMed.
- K. Effenberger, S. Breyer and R. Schobert, Eur. J. Med. Chem., 2010, 45, 1947–1954 CrossRef CAS PubMed.
- R. Schulz, T. Emmrich, H. Lemmerhirt, U. Leffler, K. Sydow, C. Hirt, T. Kiefer, A. Link and P. J. Bednarski, Bioorg. Med. Chem. Lett., 2012, 22, 6712–6715 CrossRef CAS PubMed.
- F. Zhang, X.-L. Wang, J. Shi, S.-F. Wang, Y. Yin, Y.-S. Yang, W.-M. Zhang and H.-L. Zhu, Bioorg. Med. Chem., 2014, 22, 468–477 CrossRef CAS PubMed.
- Q. P. Peterson, D. C. Hsu, D. R. Goode, C. J. Novotny, R. K. Totten and P. J. Hergenrother, J. Med. Chem., 2009, 52, 5721–5731 CrossRef CAS PubMed.
- K. S. Putt, G. W. Chen, J. M. Pearson, J. S. Sandhorst, M. S. Hoagland, J. T. Kwon, S. K. Hwang, H. Jin, M. I. Churchwell, M. H. Cho, D. R. Doerge, W. G. Helferich and P. J. Hergenrother, Nat. Chem. Biol., 2006, 2, 543–550 CrossRef CAS PubMed.
- Z. Cui, Y. Li, Y. Ling, J. Huang, J. Cui, R. Wang and X. Yang, Eur. J. Med. Chem., 2010, 45, 5576–5584 CrossRef CAS PubMed.
- S. P. Simeonov, J. A. S. Coelho and C. A. M. Afonso, ChemSusChem, 2013, 6, 997–1000 CrossRef CAS PubMed.
- S. P. Simeonov, J. A. S. Coelho and C. A. M. Afonso, ChemSusChem, 2012, 5, 1388–1391 CrossRef CAS PubMed.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- N. Shi, Q. Liu, Q. Zhang, T. Wang and L. Ma, Green Chem., 2013, 15, 1967–1974 RSC.
- B. R. Caes, M. J. Palte and R. T. Raines, Chem. Sci., 2013, 4, 196–199 RSC.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- S. Subbiah, S. P. Simeonov, J. M. S. S. Esperanca, L. P. N. Rebelo and C. A. M. Afonso, Green Chem., 2013, 15, 2849–2853 RSC.
- E. S. Kang, D. W. Chae, B. Kim and Y. G. Kim, J. Ind. Eng. Chem., 2012, 18, 174–177 CrossRef CAS PubMed.
- F. M. F. Santos, J. N. Rosa, V. André, M. T. Duarte, L. F. Veiros and P. M. P. Gois, Org. Lett., 2013, 15, 1760–1763 CrossRef CAS PubMed.
- R. S. Reddy, J. N. Rosa, L. F. Veiros, S. Caddick and P. M. P. Gois, Org. Biomol. Chem., 2011, 9, 3126–3129 CAS.
- J. N. Rosa, R. S. Reddy, N. R. Candeias, P. M. S. D. Cal and P. M. P. Gois, Org. Lett., 2010, 12, 2686–2689 CrossRef CAS PubMed.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental procedures, data and copies of 1H and 13C NMR spectra for compounds 3, 12–26 is available in supporting information. See DOI: 10.1039/c4ra03710c |
| This journal is © The Royal Society of Chemistry 2014 |