 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBorax catalyzed domino reactions: synthesis of highly functionalised pyridines, dienes, anilines and dihydropyrano[3,2-c]chromenes†
Aniruddha
Molla
and
Sahid
Hussain
*
Department of Chemistry, Indian Institute of Technology Patna, Patna 800 013, India. E-mail: sahid@iitp.ac.in; Fax: +91-612-227-7383; Tel: +91-612-255-2022
First published on 9th June 2014
Abstract
Borax, an innocuous, inexpensive, and a naturally occurring material, very efficiently catalyzes the Knoevenagel condensation and Michael addition in domino fashion for the construction of highly functionalised pyridines, dienes, anilines and dihydropyrano[3,2-c]chromenes. The present protocol offers advantages in terms of higher yields, wide scope of substrates, operational simplicity, short reaction time, no requirement of workup or column chromatography, and easy access to a wide range of structurally diverse functionalized molecules of biological importance. Recycling of the catalyst and scaling up of the reactions are important attributes of this catalytic process.
Introduction
The construction of several bonds in a one-step operation has become one of the most important tools for organic synthesis.1 It has gradually gained interest because of its remarkable advantages such as high bond forming efficiency, molecular diversity, a reduction of work-up, extraction, and purification processes, and hence minimizing waste generation making the process green.2 Therefore, researchers are endeavouring towards the construction of C–C, C–N and C–S bonds in one pot process either using one type of reaction with several reaction sites or combining more than one reaction. One of the most utilized reactions in this fashion is Knoevenagel condensation whose products (i.e. α,β-unsaturated compounds) are useful intermediates for other reactions. For instance, Knoevenagel condensation has been successfully used in the synthesis of highly functionalised molecules in combination with Michael addition, Robinson annulations, Diels–Alder and ene reactions.3The synthesis of highly functionalized pyridines, dienes, anilines and dihydropyrano[3,2-c]chromene is important as these compounds are constructed through the Knoevenagel condensation and Michael addition reactions. The adduct 3,5-dicyanopyridines are privileged heterocyclic scaffolds (Fig. 1). Modification of the substituent at C2, C4, and C6 of the pyridine core produces libraries of compounds with diverse biological activities.4 Assessment of the literature has revealed that the highly substituted pyridines and chromenes, such as those involving the 2-amino-4-aryl-6-sulfanyl substitution pattern and substituted pyrano[3,2-c]chromenes, have been particularly fruitful. These compounds find applications as antibacterial,5 anticancer,6 antiprion,7 and anti HBV infection8 compounds. Moreover, these compounds have also been used as non-nucleoside agonist of the human adenosine A19 receptor and as anti HIV-1 agents.10 Consequently, researchers have made considerable effort towards their synthesis. Traditionally, highly functionalized pyridine derivatives are prepared by refluxing active methylene compounds (malononitrile) with an aldehyde and thiols using Et3N or DABCO as a catalyst.11 Low yields (20–48%) and byproduct (enaminonitriles) formation are the main limitations of this method. In order to overcome these limitations, a few new methods have been developed using different catalysts such as nanocrystalline magnesium oxide,12 silica nanoparticles,13 KF/alumina,14 basic ionic liquid [bmIm]OH,15 ZnCl2,16 piperidine/microwave,17 DBU,18 Zn(II) and Cd(II) metal–organic frameworks (MOFs).19 Recently, KOH has also been employed for the synthesis of highly functionalized pyridine derivatives.20 Each methodology has some advantages over the others; however, the search still continues for better catalysts which bring about the Knoevenagel condensation and Michael addition simultaneously. Notably, most of the methods recently developed have involved catalysts which are air/moisture sensitive, toxic, expensive, require special effort for their preparation and special work-up and separation techniques for their recovery and recycling. Considering all these and being intrigued by the fact that the pH of an aqueous solution of borax is 9.5 and the properties of borax to produce a hydroxyl anion (Brønsted base) and boric acid (Lewis acid) upon hydrolysis that are conducive for both the Knoevenagel and Michael addition reactions,21a–d we thought it worthwhile to extend the use of borax as a potential catalyst21 to carry out the above mentioned transformations. We decided to investigate the efficacy of borax as catalyst for the synthesis of 2-amino-4-(aryl)-6-(alkyl or aryl)-3,5-pyridinedicarbonitriles, highly substituted dienes and dihydropyrano[3,2-c]chromene derivatives. Herein we report our results of borax catalyzed one-pot condensation of aldehydes, alkyl nitriles (malononitrile) and thiols (aromatic and aliphatic) for the synthesis of pyridine derivatives, cyclic monoketones for the synthesis of 1,3 diene derivatives and finally 4-hydroxycoumarin, for the synthesis of dihydropyrano[3,2-c]chromene heterocycles.
 | ||
| Fig. 1 Potent biologically active highly substituted pyridines(1–4) and naturally occurring pyranocoumarins(5). | ||
Results and discussion
In search of better catalytic systems for this domino reaction, optimization of the reaction conditions was carried out taking benzaldehyde (1.0 mmol), malononitrile (2.1 mmol) and thiophenol (1.0 mmol) as model substrates. The results of the optimization are summarized in Table 1. To initiate our study, the model reaction was performed at room temperature in the absence of catalyst; no desired product was formed both in ethanol and water. Then we studied the reaction under reflux conditions with 1 mol% of catalyst; 40% of the product was obtained in 2 hours. We increased the amount of catalyst to 5 mol% and 64% of the product was observed under the reflux conditions. We further increased the catalyst loading to 10 and 20 mol% to check the alacrity in both cases. A higher catalyst percentage did not have much effect towards decreasing the reaction time and increasing product formation. The best result was obtained with 10 mol% of catalyst in EtOH under reflux conditions (Table 2).|
|
|||||
|---|---|---|---|---|---|
| Run | Catalyst (mol%) | Solvent | Condition | Time (h) | Yielda (%) |
| a Isolated yield. | |||||
| 01 | — | H2O | R.T | 10 | — |
| 02 | — | EtOH | R.T | 10 | — |
| 03 | — | EtOH | Reflux | 10 | Trace |
| 04 | 1 | EtOH | Reflux | 2.0 | 40 |
| 05 | 5 | EtOH | Reflux | 2.0 | 64 |
| 06 | 10 | H2O | Reflux | 2.0 | 69 |
| 07 | 10 | H2O | Reflux | 1.5 | 65 |
| 08 | 10 | EtOH | R.T | 2.0 | 83 |
| 09 | 10 | EtOH | Reflux | 1.0 | 88 |
| 10 | 20 | EtOH | Reflux | 1.0 | 88 |
|
|
|||||
|---|---|---|---|---|---|
| Run | Aldehyde | Thiol | Product | Time (h) | Yielda (%) |
| a Isolated yield. b Yield after 5th run. c Yield on a 10 mmol scale. | |||||
| 01 |

|

|

|
1.0 | 88 |
| 02 |

|
|

|
1.0 | 92, 87b, 96c |
| 03 |

|

|
|
1.0 | 90 |
| 04 |

|
|
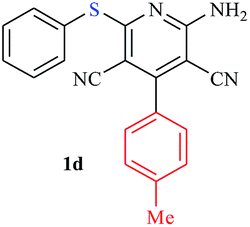
|
1.0 | 88 |
| 05 |

|

|

|
1.5 | 82 |
| 06 |
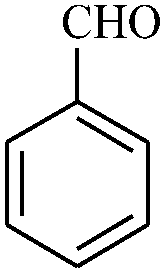
|

|

|
1.5 | 86 |
| 07 |

|

|

|
1.0 | 84 |
| 08 |
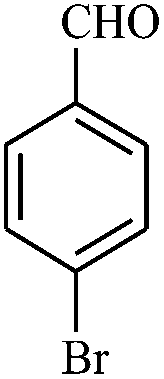
|

|
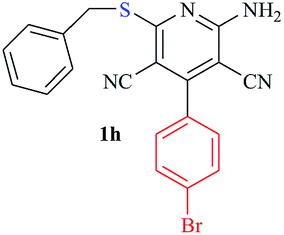
|
1.0 | 83 |
| 09 |

|

|

|
1.0 | 84 |
| 10 |
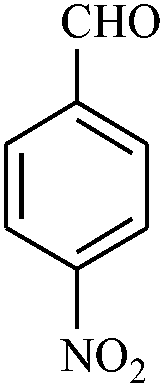
|
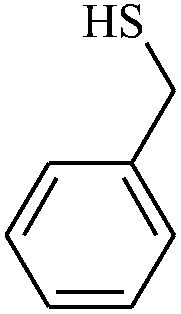
|
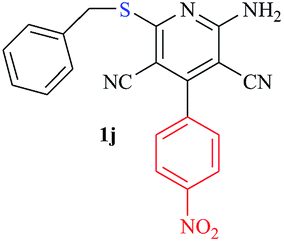
|
2.5 | 81 |
| 11 |

|

|

|
1.0 | 86 |
| 12 |
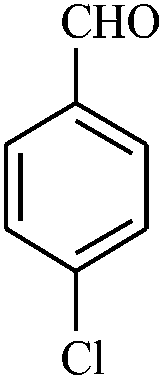
|

|
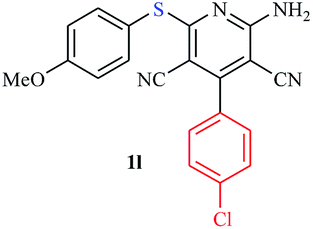
|
0.5 | 91 |
| 13 |

|

|

|
0.5 | 90 |
| 14 |

|

|

|
2.0 | 80 |
| 15 |

|

|

|
2.0 | 78 |
| 16 |

|
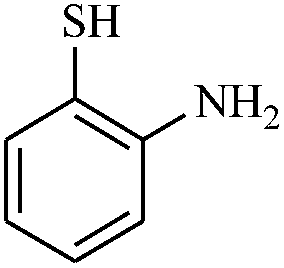
|

|
1.5 | 82 |
| 17 |

|
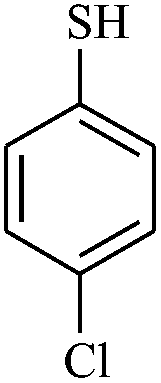
|

|
1.5 | 85 |
| 18 |
|

|

|
1.0 | 83 |
| 19 |

|

|

|
1.0 | 75 |

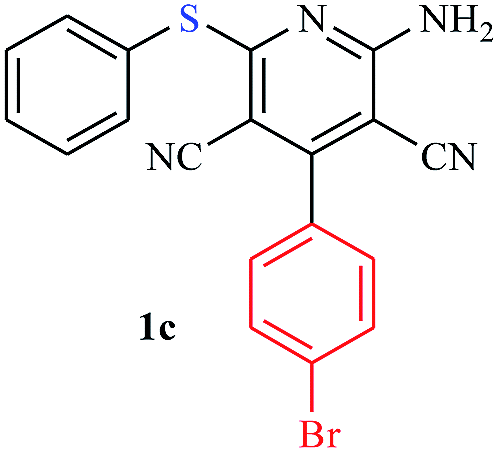
The reaction strategy worked well. Structurally diverse aromatic aldehydes bearing substituents such as Me, NO2, OMe, Cl and Br were examined with malononitrile and thiols (aliphatic and aromatic) under the optimized reaction conditions and the results presented in Table 2 (entries 1–19). The reactions with benzyl thiol went on well affording the products in high yields (Table 2, entries 6–11). Aromatic aldehydes bearing electron-withdrawing groups afforded the corresponding product with slightly longer reaction times (Table 2, entries 5 and 10). Under similar experimental conditions, aliphatic and aromatic dithiols underwent the reaction giving good yields (Table 2, entries 18 and 19). These reactions were nearly as facile as observed with the monothiols. Incidentally, reactions involving dithiols appear to be quite scarce.11b Such reactions may be highly useful in the designed synthesis of supramolecular architectures, organosulfur polymers and macromolecules. Moreover, the reactions of 2,6-dichlorobenzaldehyde afforded dihydropyridine (Table 2, entry 15). This may be due to the steric effect of two –Cl substituents in the ortho position, which prevents aromatization. We have also tried reactions with ethyl 2-cyanoacetate and 2-cyanoacetamide; only the Knoevenagel condensation product was observed with the corresponding aldehyde.
Interestingly, when we performed the reaction of 2-amino-thiophenol with malononitrile and 4-cholorobenzaldehyde, we isolated only 2-amino-6-(2-aminophenylthio)-4-(4-chlorophenyl) pyridine-3,5-dicarbonitrile as the product in good yield (Table 2, entry 16). For the same set of reactions, 2-amino-4-(4-chlorophenyl)-6-(2-mercaptophenylamino) pyridine-3,5 dicarbonitrile was also reported,20 which clearly indicates the selectivity of this protocol only with thiols over amines.
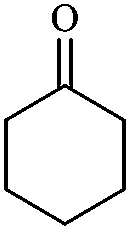
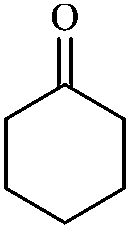
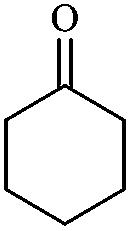
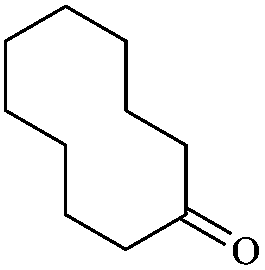
In order to extend the scope of this methodology, we carried out reactions of an aldehyde, malononitrile and enolizable ketones. It is anticipated and is evident from the literature that the combination of the above reactants in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 usually provides 2,6-dicyanoaniline derivatives. However, we could isolate diene intermediates in the case of cyclohexanone and cyclododecanone. Recently, Mukhopadhyay et al. have shown that the formation of 2,6-dicyanoaniline (i.e. aromatization) from the intermediate diene is temperature dependent.22 The synthesis of cyclohexa-1,3-diene is relatively rare22 and such wide range of functionalized dienes may find use as intermediates for the construction of larger molecules or as a relatively non-toxic precursor of HCN. Interestingly, our preliminary investigation on the above reaction produces the diene as the sole product in one hour under reflux conditions. No change in the product was observed on prolonging the reaction time (6 h). This may be due to the mild basicity of the catalyst. The reactions of other substituted aromatic aldehydes (such as Cl, Me, Br, and OMe) with cyclohexanone and malononitrile afforded the desired products in good yields (Table 3, entries 1–4). It is interesting to note that the reaction of cycloheptanone under similar reaction conditions with some selected aldehydes and malononitrile yielded aniline (and not the diene) as the sole product (Table 3, entries 5 and 6) whereas cyclododecanone produces the diene product only (Table 3, entry 7). When we tried the reaction with an acyclic ketone (butan-2-one) it failed to produce the desired product and the product of the Knoevenagel condensation was isolated. Similarly, when we changed malononitrile with ethyl 2-cyanoacetate or 2-cyanoacetamide, keeping the other reactants the same, the protocol did not work.
:2:1 usually provides 2,6-dicyanoaniline derivatives. However, we could isolate diene intermediates in the case of cyclohexanone and cyclododecanone. Recently, Mukhopadhyay et al. have shown that the formation of 2,6-dicyanoaniline (i.e. aromatization) from the intermediate diene is temperature dependent.22 The synthesis of cyclohexa-1,3-diene is relatively rare22 and such wide range of functionalized dienes may find use as intermediates for the construction of larger molecules or as a relatively non-toxic precursor of HCN. Interestingly, our preliminary investigation on the above reaction produces the diene as the sole product in one hour under reflux conditions. No change in the product was observed on prolonging the reaction time (6 h). This may be due to the mild basicity of the catalyst. The reactions of other substituted aromatic aldehydes (such as Cl, Me, Br, and OMe) with cyclohexanone and malononitrile afforded the desired products in good yields (Table 3, entries 1–4). It is interesting to note that the reaction of cycloheptanone under similar reaction conditions with some selected aldehydes and malononitrile yielded aniline (and not the diene) as the sole product (Table 3, entries 5 and 6) whereas cyclododecanone produces the diene product only (Table 3, entry 7). When we tried the reaction with an acyclic ketone (butan-2-one) it failed to produce the desired product and the product of the Knoevenagel condensation was isolated. Similarly, when we changed malononitrile with ethyl 2-cyanoacetate or 2-cyanoacetamide, keeping the other reactants the same, the protocol did not work.








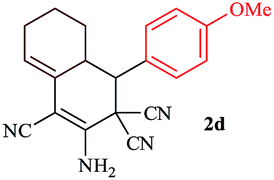
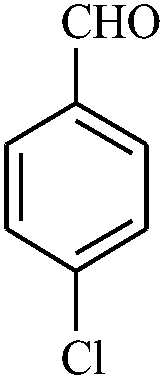



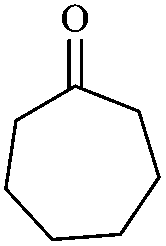
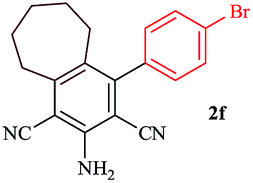


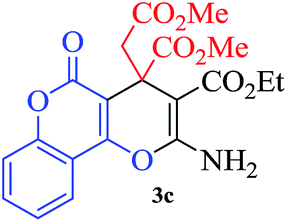
In continuation of our sustained interest in exploring the catalytic activity of borax for diverse reactions, we turned our attention towards the synthesis of dihydropyrano[3,2-c]chromene derivatives. In order to test the efficacy, we carried out reactions of equimolar amounts of 4-hydroxycoumarin, dimethylacetylenedicarboxylate (DMAD) and malononitrile in presence of 10 mol% of borax in water. Interestingly, we get 87% yield of our expected product in 1.5 h. To generalize the scope of the reaction, a series of compound (3a–f) were synthesized varying the three reactants. It was observed that the desired product was obtained in each case with 83–87% yield (Table 4, entries 1–6). It is important to note that in all the reactions the products precipitate out as a solid, making the separation of catalyst and purification of product very simple.











Recyclability of the catalyst was examined through a series of reactions with 4-chlorobenzaldehyde, malononitrile and thiophenol by using the ethanolic phase containing borax, obtained after filtration of the reaction mixture. The catalyst could be reused for at least five reaction cycles with consistent activity (Table 2, entry 2). Importantly, the reaction can be performed on a relatively large scale (10 mmol) to give good yields (Table 2, entry 2), which shows its potential for scale-up applications.
In conclusion, we have developed a facile, convenient and efficient one-pot domino reaction for the synthesis of highly substituted pyridines, dienes and chromenes of potential synthetic and pharmacological interest. This protocol offers multifarious advantages such as lower cost, a ready available and relatively non-toxic catalyst, wide scope of substrates, operational simplicity, high yields of the products and the avoidance of column chromatography. Moreover, borax, being a naturally occurring material, soluble and capable of functioning efficiently in ethanol/water, satisfies some the tenets of “green chemistry.” The reactions are thus more conducive to the environment and provide scope for further study.
Synthesis of 2-amino-4-(aryl)-6-(alkyl or aryl)-3,5-pyridinedicarbonitriles
To a solution of aldehyde (1.0 mmol), malononitrile (2.1 mmol) and borax (0.1 mmol) in 3 mL of ethanol was added thiols (1.0 mmol) at room temperature in a round bottom flask equipped with a condenser. The reaction mixture was stirred under reflux conditions for the required time (Table 2) and then allowed to cool to room temperature. The progress of the reaction was monitored by TLC, and after completion of the reaction, the reaction mixture was gradually cooled to room temperature. The solid product was filtered off, washed with ethanol and dried to obtain the corresponding product.Synthesis of dienes and aniline derivatives
To a solution of aldehyde (1.0 mmol), malononitrile (2.1 mmol) and borax (0.1 mmol) in 3 mL of ethanol was added the cyclic ketone (1.0 mmol) at room temperature in a round bottom flask equipped with a condenser. The reaction mixture was stirred under reflux conditions for the required time (Table 3) and then allowed to cool to room temperature. The progress of the reaction was monitored by TLC, and after completion of the reaction, the reaction mixture was gradually cooled to room temperature. The solid product was filtered off, washed with ethanol and dried to obtain the corresponding product.(E)-2-Amino-4-(4-chlorophenyl)-4a,5,6,7,8,9,10,11-octahydrobenzo[10]annulene-1,3,3(4H)-tricarbonitrile (2g)
Yield 76%. Solid, m.p. 230–232 °C. IR νmax (KBr): 3350, 3229, 3099, 2926, 2857, 2200, 1672, 1586, 1421, 1093 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 1.29–1.45 (m, 7H), 1.57–1.63 (m, 2H), 2.15–2.20 (m, 2H), 2.51–2.53 (m, 2H), 2.79–2.83 (m, 2H), 6.23–6.37 (m, 2H), 7.28–7.30 (m, 2H), 7.34–7.36 (m, 2H), 8.17 (br s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6 + CDCl3): 28.5, 28.6, 28.7, 28.8, 28.9 (2C), 32.4, 32.9, 42.0, 76.9, 99.5, 112.7, 113.0, 127.2, 128.2, 128.3, 131.2, 131.7, 136.2, 157.6, 163.4 ppm. Anal. calcd for C20H13N5O2S: C, 62.00; H, 3.38; N, 18.08; obtained C, 61.96; H, 3.36; N, 18.11%.Synthesis of dihydropyrano[3,2-c]chromene derivatives
To a solution of 4-hydroxy derivative (1.0 mmol), malononitrile (1.1 mmol) and borax (0.1 mmol) in 3 mL of water was added dimethyl/diethyl acetylenedicarboxylate (1.0 mmol) at room temperature in a round bottom flask equipped with a condenser. The reaction mixture was stirred under reflux conditions for the required time (Table 4) and then allowed to cool to room temperature. The progress of the reaction was monitored by TLC, and after completion of the reaction, the reaction mixture was gradually cooled to room temperature. The solid product was filtered off, washed with ethanol and dried to obtain the corresponding product.Acknowledgements
A.M. is thankful to IIT Patna for his research fellowship. The authors are thankful to SAIF (Panjab University, Chandigarh) and IIT Kanpur for NMR facility. The authors are also thankful to the Department of Science and Technology for research grant no. SR/FT/CS-093/2009.References
- (a) L. F. Tietze, G. Brasche and K. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 1996 Search PubMed; (b) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed; (c) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef; (d) J. Zhu and H. BienayméMulticomponent Reactions, 1st edn, 2005 Search PubMed.
- (a) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed; (b) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed; (c) B. B. Toure and D. G. Hall, Chem. Rev., 2009, 109, 4439–4486 CrossRef CAS PubMed; (d) B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS PubMed; (e) B. Jiang, T. Rajale, W. Wever, S.-J. Tu and G. Li, Chem.–Asian J., 2010, 5, 2318–2335 CrossRef CAS PubMed; (f) Y. Huang, A. Yazbak and A. Dömling, Multicomponent Reactions, in Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. W. Cue, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed; (g) A. Doling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (h) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS PubMed; (i) A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef CAS PubMed.
- (a) D. B. Ramachary and M. J. Kishor, J. Org. Chem., 2007, 72, 5056–5068 CrossRef CAS PubMed; (b) D. B. Ramachary, K. Ramakumar and V. V. Narayana, J. Org. Chem., 2007, 72, 14581 Search PubMed; (c) A. Kumar and R. A. Maurya, Tetrahedron, 2007, 63, 1946–1952 CrossRef CAS PubMed; (d) G. Sabitha, N. Fatima, E. V. Reddy and J. S. Yadav, Adv. Synth. Catal., 2005, 347, 1353–1355 CrossRef CAS; (e) D. B. Ramachary, K. Anebouselvy, N. S. Chowdari and C. F. Barbas, III, J. Org. Chem., 2004, 69, 5838–5849 CrossRef CAS PubMed; (f) G. Sabitha, G. S. K. K. Reddy, M. Rajkumar, J. S. Yadav, K. V. S. Ramakrishna and A. C. Kunwar, Tetrahedron Lett., 2003, 44, 7455–7457 CrossRef CAS PubMed; (g) P. A. Clarke and W. H. C. Martin, Org. Lett., 2002, 4, 4527–4529 CrossRef CAS PubMed; (h) C. Tratrat, S. Giorgi-Renault and H.-P. Husson, Org. Lett., 2002, 4, 3187–3189 CrossRef CAS PubMed; (i) S. Marchalin, K. Cvopova, D. P. Pham-Huu, M. Chudik, J. Kozisek, I. Svoboda and A. Daeich, Tetrahedron Lett., 2001, 42, 5663–5667 CrossRef CAS; (j) J. M. Betancort, K. Sakthivel, R. Thayumanavan and C. F. Barbas, III, Tetrahedron Lett., 2001, 42, 4441–4444 CrossRef CAS; (k) L. F. Tietze and Y. Zhou, Angew. Chem., Int. Ed., 1999, 38, 2045–2047 CrossRef CAS; (l) M. Miyano and M. A. Stealey, J. Org. Chem., 1982, 47, 3184–3186 CrossRef CAS.
- M. T. Cocco, C. Congiu, V. Lilliu and V. Onnis, Eur. J. Med. Chem., 2005, 40, 1365–1372 CrossRef CAS PubMed and references cited therein.
- (a) S. K. Srivastava, R. P. Tripathi and R. Ramachandran, J. Biol. Chem., 2005, 280, 30273–30281 CrossRef CAS PubMed; (b) H. Brötz-Oesterhelt, I. Knezevic, S. Bartel, T. Lampe, U. Warnecke-Eberz, K. Ziegelbauer, D. Häbich and H. Labischinski, J. Biol. Chem., 2003, 278, 39435–39442 CrossRef PubMed.
- M. A. Azuine, H. Tokuda, J. Takayasu, F. Enjyo, T. Mukainaka, T. Konoshima, H. Nishino and G. J. Kapadia, Pharmacol. Res., 2004, 49, 161–169 CrossRef CAS PubMed.
- T. R. K. Reddy, R. Mutter, W. Heal, K. Guo, V. J. Gillet, S. Pratt and B. Chen, J. Med. Chem., 2006, 46, 607–615 CrossRef PubMed.
- H. Chen, W. Zhang, R. Tam and A. K. Raney, PCT Int. Appl. WO 2005058315 A1 20050630, 2005.
- L. H. Heitman, T. Mulder-Krieger, R. F. Spanjersberg, J. K. von Frijtag Drabbe Künzel, A. Dalpiaz and A. P. Ijzerman, Br. J. Pharmacol., 2006, 147, 533–541 CrossRef CAS PubMed.
- J. Deng, T. Sanchez, L. Q. Al-Mawsawi, R. Dayam, R. A. Yunes, A. Garofalo, M. B. Bolger and N. Neamati, Bioorg. Med. Chem., 2007, 15, 4985–5002 CrossRef CAS PubMed.
- (a) N. M. Evdokimov, I. V. Magedov and A. S. Kireev, Org. Lett., 2006, 8, 899–902 CrossRef CAS PubMed; (b) N. M. Evdokimov, A. S. Kireev, A. A. Yakovenko, M. Y. Antipin, I. V. Magedov and A. Kornienko, J. Org. Chem., 2007, 72, 3443–3453 CrossRef CAS PubMed.
- M. L. Kantam, K. Mahendar and S. Bhargava, J. Chem. Sci., 2010, 122, 63–69 CrossRef CAS PubMed.
- S. Banerjee and G. Sereda, Tetrahedron Lett., 2009, 50, 6959–6962 CrossRef CAS PubMed.
- K. N. Singh and S. K. Singh, ARKIVOC, 2009, xiii, 153–160 CrossRef.
- B. C Ranu, R. Jana and S. Sowmiah, J. Org. Chem., 2007, 72, 3152–3154 CrossRef PubMed.
- M. Sridhar, B. C. Ramanaiah, C. Narsaiah, B. Mahesh, M. Kumaraswamy, K. K. R. Mallu, V. M. Ankathi and P. S. Rao, Tetrahedron Lett., 2009, 50, 3897–3900 CrossRef CAS PubMed.
- K. Guo, R. Mutter, W. Heal, T. R. K. Reddy, H. Cope, S. Pratt, M. J. Thompson and B. Chen, Eur. J. Med. Chem., 2008, 43, 93–106 CrossRef CAS PubMed.
- R. Mamgain, R. Singh and D. S. Rawat, J. Heterocycl. Chem., 2009, 46, 69–73 CrossRef CAS.
- M. Thimmaiah, P. Li, S. Regati, B. Chen and J. C.-G. Zhao, Tetrahedron Lett., 2012, 53, 4870–4872 CrossRef CAS PubMed.
- M. N. Khan, S. Pal, T. Parvin and L. H. Choudhury, RSC Adv., 2012, 2, 12305–12314 RSC.
- (a) A. Molla, E. Hossain and S. Hussain, RSC Adv., 2013, 3, 21517–21523 RSC; (b) S. Hussain, S. K. Bharadwaj, R. Pandey and M. K. Chaudhuri, Eur. J. Org. Chem., 2009, 3319–3322 CrossRef CAS; (c) S. Hussain, S. K. Bharadwaj, M. K. Chaudhuri and H. Kalita, Eur. J. Org. Chem., 2007, 374–378 CrossRef CAS; (d) M. K. Chaudhuri, S. Hussain, M. L. Kantam and B. Neelima, Tetrahedron Lett., 2005, 46, 8329–8331 CrossRef CAS PubMed; (e) M. K. Chaudhuri and S. Hussain, J. Mol. Catal. A: Chem., 2005, 269, 214–217 CrossRef PubMed.
- P. Das, R. J. Butcher and C. Mukhopadhyay, Green Chem., 2012, 14, 1376–1387 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of all compounds are available. See DOI: 10.1039/c4ra03627a |
| This journal is © The Royal Society of Chemistry 2014 |