
An efficient and new protocol for phosphine-free Suzuki coupling reaction using palladium-encapsulated and air-stable MIDA boronates in an aqueous medium
Abstract
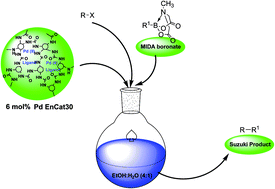
A simple methodology that uses a system based on polyurea microencapsulated palladium (PdEnCat 30™) and aryl or (2-pyridyl) MIDA boronates for Suzuki–Miyaura cross-coupling reactions of (hetero)aryl halides in water–alcohol under phosphine-free conditions was developed.
 Please wait while we load your content...
Please wait while we load your content...

