
Reduced recombination and enhanced UV-assisted photocatalysis by highly anisotropic titanates from electrospun TiO2–SiO2 nanostructures†
Abstract
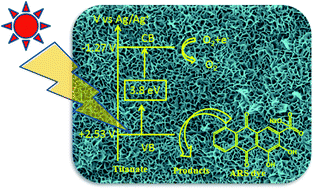
The surface areas of electrospun fibers/rice grain-shaped nanostructures of TiO2–SiO2 composites were further enhanced after transforming them into thorn or sponge shaped titanates via selective leaching of SiO2, which was reported by our group previously [RSC Adv., 2012, 2, 992]. In this study, we report on their application in photocatalytic activity (PCA) when juxtaposed with photoluminescence (PL). Two defect related bands are observed in PL and their origin is discussed in relation to calcination, crystallization and nucleation effects. The relative PL intensity for sponge shapes was the lowest and hence had the lowest radiative recombination, which suggests carrier trapping at defect centers. This enables the charge carriers to migrate to the surface and participate in the PCA. The results of PCA suggested that the sponge-shaped titanate exhibits the highest degradation rate among all samples. A plausible mechanism for the differences in PCA is proposed based on the variation in the defect-densities.
 Please wait while we load your content...
Please wait while we load your content...

