 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA “green” variation of the Hirao reaction: the P–C coupling of diethyl phosphite, alkyl phenyl-H-phosphinates and secondary phosphine oxides with bromoarenes using a P-ligand-free Pd(OAc)2 catalyst under microwave and solvent-free conditions†
György
Keglevich
*,
Erzsébet
Jablonkai
and
László B.
Balázs
Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary. E-mail: gkeglevich@mail.bme.hu; Fax: +36 1 463 3648; Tel: +36 1 463 1111/5883
First published on 8th May 2014
Abstract
The P–C coupling of diethyl phosphite, alkyl phenyl-H-phosphinates, diphenylphosphine oxide and dialkylphosphine oxides with bromoarenes may be performed in the presence of a P-ligand-free Pd(OAc)2 catalyst and triethylamine under microwave-assisted (MW) and, in almost all cases, solvent-free conditions to afford diethyl arylphosphonates, alkyl diphenylphosphinates, aryldiphenylphosphine oxides and dialkylphenylphosphine oxides, respectively. This is the “greenest” accomplishment of the well-known Hirao reaction that has now been found to have general application for a broad spectrum of >P(O)H species with different reactivity and a great variety of substituted bromobenzenes. The alkyl phenyl-H-phosphinates were prepared by the MW-promoted alkylation of phenyl-H-phosphinic acid in the absence of any solvent.
Introduction
The synthesis of aryl phosphonates and related derivatives is a focus of interest these days.2 The preparation of aryl phosphonates by the Arbuzov reaction of trialkyl phosphites and aryl halides is possible only under special conditions due to the lower reactivity of aryl halides.3,4 The most suitable method for the synthesis of aryl phosphonates is the Hirao reaction comprising a P–C coupling between a dialkyl phosphite (H-phosphonate) and an aryl or vinyl halide (or another aryl derivative) in the presence of Pd(PPh3)4 as the catalyst, and in most cases, triethylamine as the base in different solvents.5–12 The first examples of the Hirao reaction included also the preparation of vinyl phosphates from vinyl halides. Many variations and applications of the Hirao reaction have been described; the coupling was extended to H-phosphinates, secondary phosphine oxides and other P-reactants, as well as to other Pd(0) complexes and Pd(II) salts applied in the presence of suitable P-ligands.2 According to one method, the P–C coupling was enhanced by microwave (MW) irradiation, but this method did not bring a breakthrough, as Pd(PPh3)4 and THF had to be used.13 In another case,14 MW irradiation also had only an accelerating effect. It was a milestone, when attempts were made to replace Pd(PPh3)4 with Pd(dba)2 (dba = dibenzylideneacetone), Pd(OAc)2 or PdCl2. The most efficient catalytic system was formed from Pd(OAc)2 in the presence of triphenylphosphine as the P-ligand.15 There have been attempts to apply bidentate P-ligands, such as dppf [1,1′-bis(diphenylphosphino)ferrocene], xantphos [4,5-bis(diphenylphosphino)-9,9-dimethylxanthene], dppp [1,3-bis(diphenylphosphino)propane], dppb [1,4-bis(diphenylphosphino)butane], and BINAP [2,2′-bis(diphenylphosphino)-1,1′-binaphthyl] together with Pd(OAc)2, instead of triphenylphosphine, to establish in situ catalysts.16–18 In another variation, a Pd(II) salt was used with triphenylphosphine, but the base was K2CO3, and the reaction was performed in the presence of triethylbenzylammonium chloride (TEBAC) as the phase transfer catalyst.19–22 It was observed that the phosphinylation of aryl iodides took place in the presence of ‘phosphine-free Pd’, but aryl bromides underwent the coupling reaction only in the presence of triphenylphosphine as the P-ligand.19–22 Diphenylphosphine may also be the subject of an analogous coupling reaction using Pd(OAc)2 catalyst under MW irradiation. However, this kind of P–C coupling has been limited only to iodobenzene as the reactant.23 The high ability of Ph2PH towards oxidation means another disadvantage, and this reaction was not described as a general method. It is noteworthy that reductive Hirao couplings were also described using NiBr2 and Mg,24 or NiCl2 and Zn along with 2,2′-bipyridine and K3PO4 in a suitable solvent.25We experienced that in certain alkylation reactions, the catalysts could be substituted by MW irradiation,26–30 or it influenced positively the effect of a catalyst.4,31,32 For this reason, we wished to investigate the role of MW irradiation in the Hirao reaction. We envisaged that the catalyst system may be simplified. It was a question of whether it might be possible to omit the P-ligand under certain conditions in the coupling reaction of bromoarenes. If the simplification of the catalytic system is possible, for which >P(O)H reagents might it be relevant? Our preliminary results on a limited scope of the P–C coupling reactions have been published in a communication.1
Results and discussion
Our model reaction was the P–C coupling between bromoarenes and, in most cases, 1.5–1 equivalents of diethyl phosphite, alkyl phenyl-H-phosphinates and secondary phosphine oxides using Pd(OAc)2 as the catalyst without any P-ligand and 1.1 equivalents of triethylamine as the base in the absence of any solvent on MW irradiation. The basic model was the coupling of diethyl phosphite with bromoarenes (Scheme 1).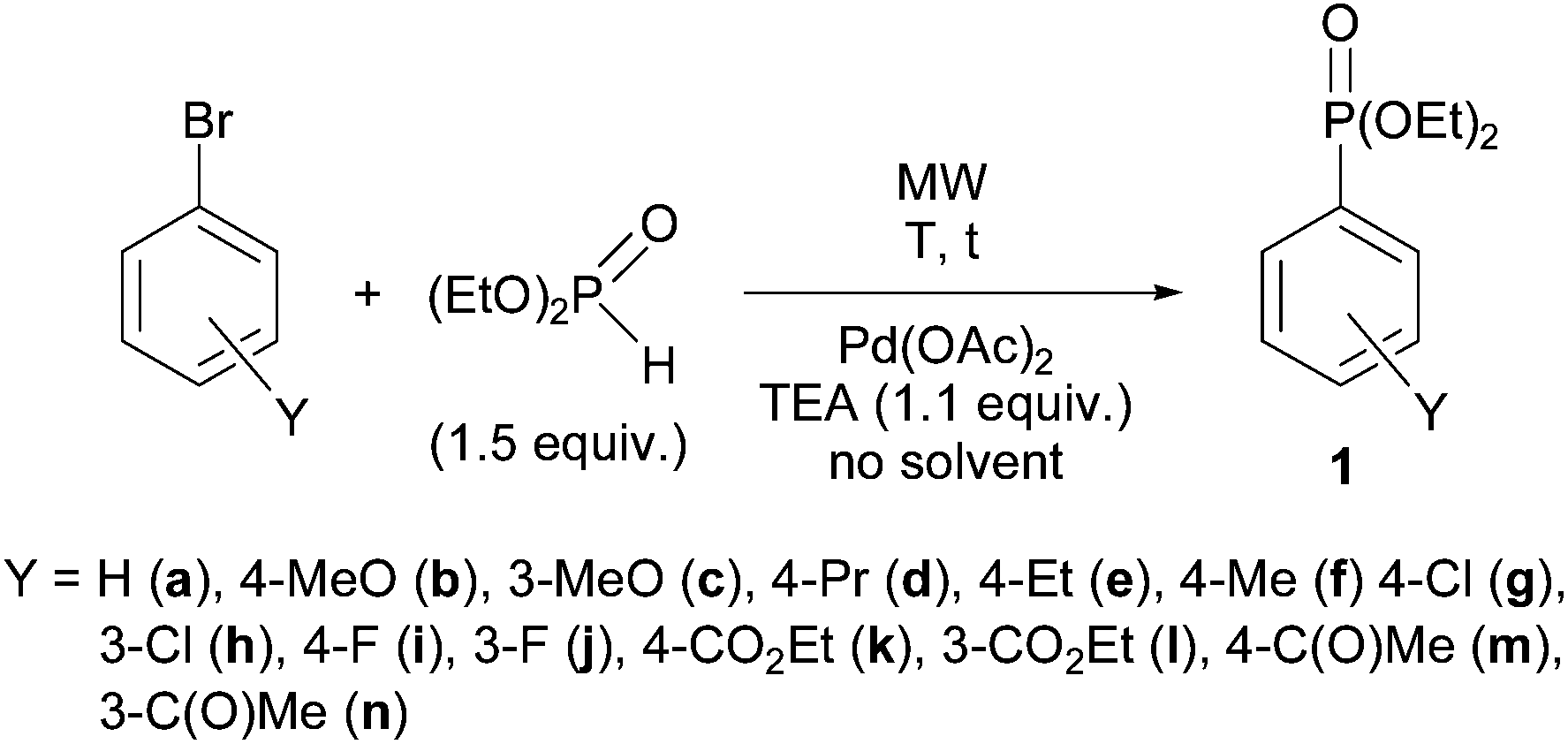 | ||
| Scheme 1 P–C coupling reaction of bromoarenes with diethyl phosphite. | ||
First, the reaction of diethyl phosphite (DEP) with bromobenzene was investigated. The coupling was complete in the presence of 5 mol% of Pd(OAc)2 at 150 °C, after an irradiation of 5 min, and diethyl phenylphosphonate 1a was obtained in a yield of 93% (Table 1, entry 1).1 The comparative thermal reaction led to a conversion of only 47% (Table 1, entry 2).
| Entry | Y | Pd(OAc)2 (%) | T (°C) | t (min) | Mode of heating | Conversiona (%) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
a On the basis of GC analysis 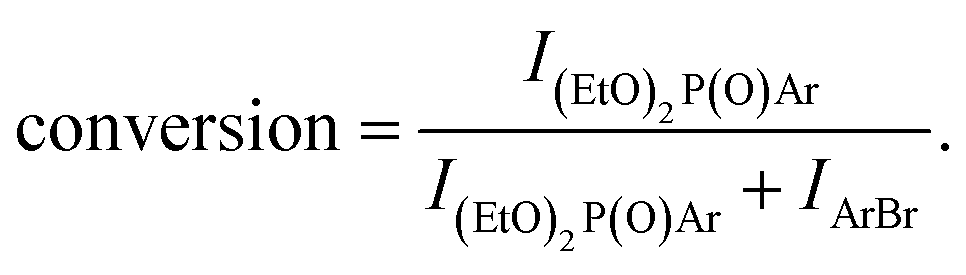 b In this case 3 equiv. DEP were used.
c Conventional heating.
d Average of two reactions. The deviation is ±1.5%.
e No change on further irradiation.
f On further irradiation, the product decomposed.
g Quasi-isothermal reaction.
h The starting material was partially.
b In this case 3 equiv. DEP were used.
c Conventional heating.
d Average of two reactions. The deviation is ±1.5%.
e No change on further irradiation.
f On further irradiation, the product decomposed.
g Quasi-isothermal reaction.
h The starting material was partially.
|
||||||||
| 1 | H | 5 | 150 | 5 | MW | 99 | 93 (1a) | 1 |
| 2 | H | 5 | 150 | 5 | Δc | 47 | 27 (1a) | |
| 3 | 4-MeOb | 5 | 150 | 10 | MW | 57d,e | 50 | |
| 4 | 4-MeO | 10 | 150 | 5 | MW | 67d,e | 56 | |
| 5 | 4-MeO | 10 | 150 | 5 | Δc | 38d | 30 | |
| 6 | 4-MeO | 10 | 150 | 15 | Δc | 62e | 53 | |
| 7 | 4-MeO | 10 | 175 | 5 | MW | 77d,f | 66 | |
| 8 | 4-MeO | 10 | 200g | 2 | MW | 80f | 69 (1b) | |
| 9 | 3-MeOb | 10 | 150 | 5 | MW | 78e | ||
| 10 | 3-MeO | 10 | 175 | 5 | MW | 81e | 73 | |
| 11 | 3-MeO | 10 | 200g | 2 | MW | 93f | 79 (1c) | |
| 12 | 4-Pr | 10 | 150 | 15 | MW | 70 | ||
| 13 | 4-Pr | 10 | 175 | 5 | MW | 77f | ||
| 14 | 4-Pr | 10 | 200g | 2 | MW | 86f | 71 (1d) | |
| 15 | 4-Et | 10 | 150 | 15 | MW | 61 | ||
| 16 | 4-Et | 10 | 175 | 15 | MW | 93f | 85 (1e) | |
| 17 | 4-Me | 10 | 175 | 10 | MW | 86f | 73 (1f) | 1 |
| 18 | 4-Cl | 10 | 175 | 10 | MW | 95 | 83 (1g) | 1 |
| 19 | 3-Cl | 10 | 150 | 10 | MW | 73e | ||
| 20 | 3-Cl | 10 | 175 | 10 | MW | 95 | 87 (1h) | |
| 21 | 3-Cl | 10 | 200g | 2 | MW | 92 | ||
| 22 | 4-F | 5 | 175 | 5 | MW | 99 | 91 (1i) | 1 |
| 23 | 3-F | 5 | 150 | 5 | MW | 70 | ||
| 24 | 3-F | 5 | 175 | 10 | MW | 100 | 88 (1j) | |
| 25 | 3-F | 10 | 175 | 5 | MW | 95 | ||
| 26 | 4-CO2Et | 5 | 175 | 15 | MW | 100 | 89 (1k) | 1 |
| 27 | 3-CO2Et | 10 | 150 | 15 | MW | 66e | ||
| 28 | 3-CO2Et | 10 | 175 | 5 | MW | 83f | 74 | |
| 29 | 3-CO2Et | 10 | 200g | 2 | MW | 93f | 81 (1l) | |
| 30 | 4-C(O)Me | 5 | 175 | 5 | MW | 96h | 71 (1m) | 1 |
| 31 | 3-C(O)Me | 10 | 150 | 15 | MW | 76 | ||
| 32 | 3-C(O)Me | 5 | 175 | 10 | MW | 81 | ||
| 33 | 3-C(O)Me | 10 | 175 | 5 | MW | 100 | 92 | |
| 34 | 3-C(O)Me | 5 | 200g | 2 | MW | 100 | 89 (1n) | |
Then, DEP was reacted with a series of substituted aryl bromides. Using 4-methoxybromobenzene at 150 °C in the presence of 5 mol% and 10 mol% Pd(OAc)2, incomplete conversions of 57% and 67%, respectively, were observed and the compositions did not change on extending the reaction times of 10/5 min (Table 1, entries 3 and 4). A comparative thermal experiment carried out in the presence of 10 mol% of Pd(OAc)2 at 150 °C for 5 min led to a conversion of only 38% (Table 1, entry 5), that was increased to 62% after a heating of 15 min, but than the conversion could not be increased further (Table 1, entry 6). Increasing the temperature to 175 °C and 200 °C, and applying reaction times of 5 min and 2 min, the conversions were 77% and 80%, respectively, and decomposition was observed on further irradiation (Table 1, entries 7 and 8). Hence, the last two experiments were the best giving diethyl arylphosphonates 1b in yields of 66% and 69%, respectively (Table 1, entries 7 and 8). It is obvious that the 4-methoxybromobenzene is significantly less reactive in the reaction under discussion than bromobenzene. The 3-methoxybromobenzene was, however, somewhat more reactive than the 4-methoxy substituted analogue, as measuring in 10 mol% of the catalyst and applying conditions of 150 °C/5 min, 175 °C/5 min and 200 °C/2 min, the conversions were 78%, 81% and 93%, respectively (Table 1, entries 9–11). The preparative yield of 3-methoxyphenylphosphonate 1c from the best experiment was 79% (Table 1, entry 11). At 150/175 °C, the conversions remained incomplete even on prolonged heating, while at 200 °C, decomposition was observed after a reaction time of 2 min. In the next experiments 4-alkyl substituted bromobenzenes were the model compounds in reaction with 1.5 equivalents of DEP. First, 4-propylbromobenzene was used as the starting material. Applying a combination of 150 °C/15 min, 175 °C/5 min and 200 °C/2 min in the presence of 10 mol% of Pd(OAc)2, the conversions were 70%, 77% and 86%, respectively (Table 1, entries 12–14). The conversions were incomplete and the prolonged heating had a negative effect on the yield. From the best experiment, the 4-propylphenylphosphonate (1d) was isolated in a yield of 71% (Table 1, entry 14). In respect of the 4-ethyl- and 4-methylbromobenzenes, a reaction temperature of 175 °C seemed to be the optimum to afford products 1e and 1f in conversions of 93% and 86%, respectively, after reaction times of 15 min and 10 min, respectively (Table 1, entries 16 and 17). The 4-ethylphenylphosphonate (1e) was isolated in a yield of 85%, while the 4-methyl counterpart (1f) in a yield of 73% (Table 1, entries 16 and 17). It can be seen that the 4-alkyl substituted bromobenzenes are also less reactive than bromobenzene. Above 175 °C, the phosphonates with electron-donating substituents in the aromatic ring (e.g.1b–f) were not entirely stable and decomposed partially to give the corresponding benzene derivative as a minor by-product.
The next experiments embraced the reactions of halogeno-substituted bromobenzenes. The coupling reactions of 4-chlorobromobenzene and the 3-chloro analogue were quite efficient in the presence of 10 mol% of the catalyst at 175 °C after an irradiation of 10 min, as chlorophenylphosphonates 1g and 1h were obtained in conversions of 95% for both cases, and in yields of 83% and 87%, respectively (Table 1, entries 18 and 20). At 150 °C, the conversion was lower (73%), while at 200 °C it remained practically the same (92%) as that detected at 175 °C (Table 1, entries 19 and 21). The fluoro-substituted bromobenzenes were the most efficient reagents after bromobenzene. Both the 4-fluoro- and the 3-fluorobromobenzene took part in a quantitative reaction with DEP in the presence of 5 mol% of Pd(OAc)2 as the catalyst at 175 °C after a reaction time of 5 min and 10 min, respectively, to furnish fluorophenylphosphonates 1i and 1j in 91% and 88% yields, respectively (Table 1, entries 22 and 24). In respect of the 3-fluoro starting material, the coupling was incomplete at 150 °C for 5 min, while using more (10 mol%) of the catalyst, the shorter reaction time of 5 min was enough (Table 1, entries 23 and 25). It was a noteworthy observation that the coupling reaction of the bromo-function of the dihalogenobenzenes with the >P(O)H species was entirely selective, the chloro- and fluoro-moieties remained intact under the MW-assisted P-ligand-free and solvent-free conditions.
The next experiments involved a study with the ethyl 4- and 3-bromobenzoates. The 4-ethoxycarbonyl- and 3-(ethoxycarbonyl)bromobenzene could be involved in quite efficient couplings using 5 mol% of Pd(OAc)2 at 175 °C for 15 min and 10 mol% of the catalyst at 200 °C for 2 min, respectively, to provide products 1k and 1l in yields of 89% and 81%, respectively (Table 1, entries 26 and 29). Regarding the reaction of ethyl 3-bromobenzoate, lower temperatures than 200 °C were less efficient (Table 1, entries 27 and 28). At 150 °C incomplete conversions occurred, while at 175 °C/200 °C decomposition of the product (1l) was observed (Table 1, entries 27–29). It can be seen that the ethyl bromobenzoates are less reactive than bromobenzene.
Finally, the 4- and 3-bromoacetophenones were tested. The coupling reaction of 4-bromoacetophenone with DEP was complete using 5 mol% of Pd(OAc)2 at 175 °C for 5 min, but a partial dehalogenation of the starting material was also observed (Table 1, entry 30). At the same time, the reaction of the 3-bromoacetophenone took place quantitatively in the presence of 10 mol% of catalyst at 175 °C after 5 min, or applying 5 mol% of catalyst at 200 °C after 2 min (Table 1/entries 33 and 34). At 150 °C/15 min in the presence of 10 mol% of Pd(OAc)2, or 175 °C/10 min using 5 mol% of the catalyst, the conversions were 76% and 81%, respectively (Table 1, entries 31 and 32). The 3-acetylbromobenzene was found to reveal a comparable reactivity with that of the 4-bromobenzoate.
It is worthy to mention that the reaction mixtures formed a homogeneous liquid phase. Due to the stirring, there could not have been a temperature gradient in the mixtures.
It was found that the addition of 10 mol% of PPh3 (1 equivalent to the catalyst) to the reaction mixture was without any effect. Repeating the experiment marked by entry 4 of Table 1 in the presence of PPh3, the yield of arylphosphonate 1b was 57%.
It can be seen that with the exception of 4-methoxybromobenzene, all bromoarenes investigated could be converted into the corresponding diethyl arylphosphonates (1) in conversions ≥86%, although the optimum conditions were somewhat different. The overall reactivity of the aromatic substrates was the following: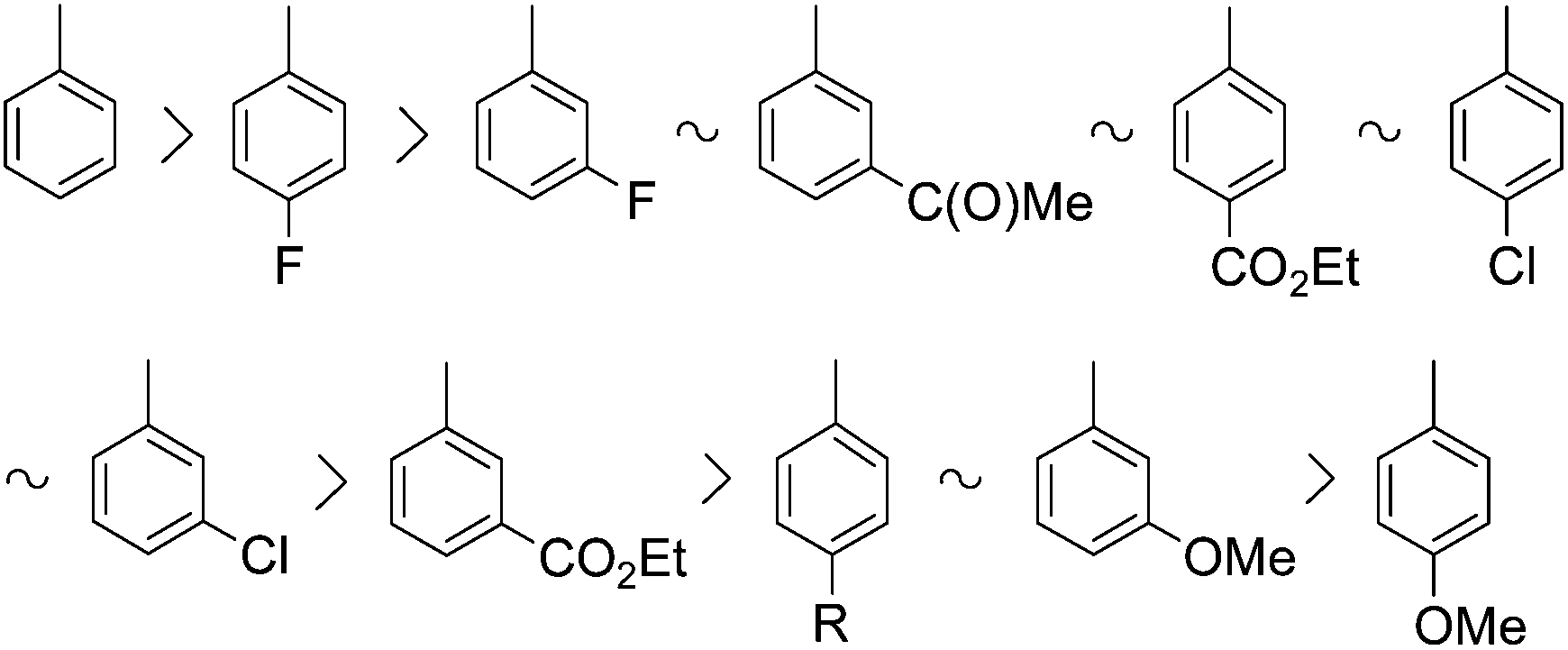
One may conclude that the presence of substituents in general decreases the reactivity of the bromoarenes and that electron-donating (methoxy and alkyl) substituents have a more significant effect in this respect than electron-withdrawing (halogeno, acetyl and ethoxycarbonyl) substituents. In the case of electron-donating substituents, the conversions were incomplete and the best yields were 69–85%. 4-Methoxybromobenzene revealed the lowest reactivity among the aromatic substrates used.
The main message of our finding is that, first in the literature, the Hirao reaction of bromoarenes could be performed in the presence of P-ligand-free Pd(OAc)2 that now proved to be general for a wide variety of substituted bromobenzenes. This was possible only under MW irradiation, as shown by the result of comparative thermal experiments. Moreover, our procedure can be carried out under solvent-free conditions. The explanation for the beneficial influence of MW irradiation may be that the statistically occurring local overheating effect promotes the P–C coupling in the absence of P-ligands.33 This experience augments the number of cases, when MW irradiation simplifies the realization of catalytic reactions.4,26–32
From among the diethyl arylphosphonates (1a–n) synthesized, 1a–c, 1e–g, 1i–k, 1m and 1n prepared are known compounds.1,3,34–39 All arylphosphonates (1) were characterized by 31P, 13C, 1H NMR, and HR-MS.
In the next part of our work, we wished to utilize alkyl phenyl-H-phosphinates in the Hirao reaction. First we had to prepare the phenylphosphinates (2). This was done essentially on the basis of our previous method involving the alkylating esterification of phosphinic acids under MW- and solvent-free conditions.40,41 According to this, phenyl-H-phosphinic acid was reacted with alkyl halides in the presence of triethylamine at 80 °C for 5–15 min with MW irradiation without the use of any solvent (Scheme 2, Table 2). In our original procedure, K2CO3 was used as the base together with a phase transfer catalyst.40,41 Now, triethylamine was applied in a homogeneous medium for the synthesis of never representatives.
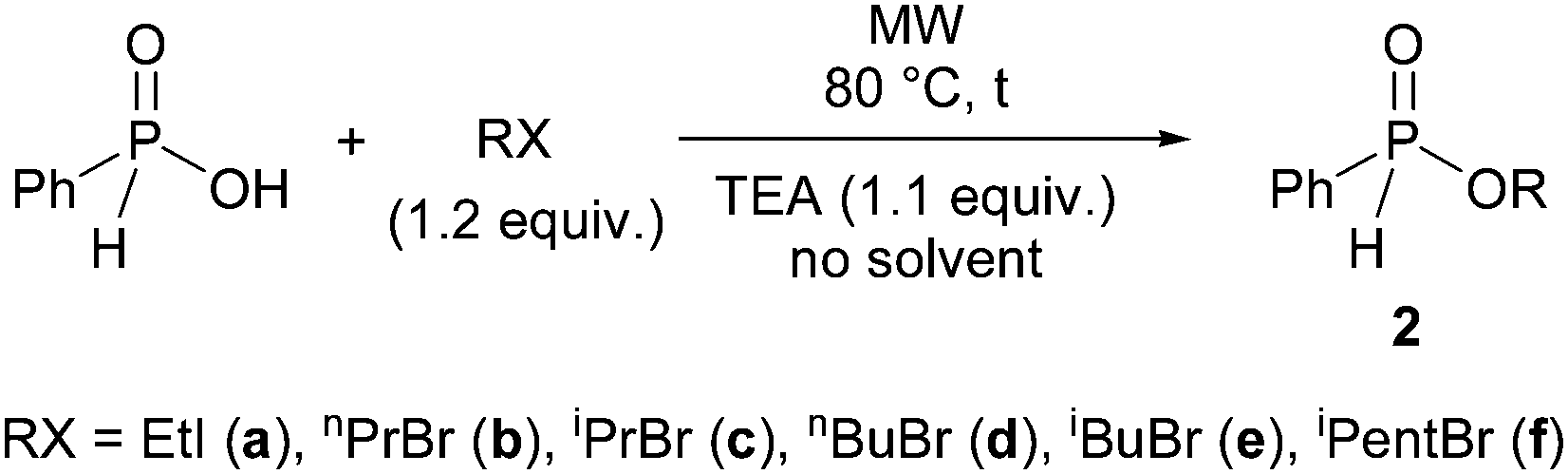 | ||
| Scheme 2 Alkylating esterification of phenyl-H-phosphinic acid with alkyl halides. | ||
One may see from Table 2 that using ethyl iodide, n-propyl bromide, isopropyl bromide, n-butyl bromide, isobutyl bromide and isopentyl bromide, the corresponding alkyl phenyl-H-phosphinates (2a–f) were obtained in yields of 76–97% after flash column chromatography. In these O-alkylation reactions the effect of MW is noteworthy, if the results are compared with those of the comparative thermal experiments. In the alkylation with isopropyl bromide at 80 °C for 15 min, the MW-assisted reaction gave phenylphosphinate 2c in a yield of 76% (Table 2, entry 3). As the same time, the outcome of the comparative thermal experiment was only 26% (Table 2, entry 4). Increasing the reaction time to 30 min, the yield was doubled (53%) (Table 2, entry 5). In the alkylation with butyl bromide, the effect of MW was also considerable, but after a prolonged reaction time, almost the same yield (94%) could be achieved on conventional heating, as that in the MW variation (96%) (Table 2, entries 6–8). From among the H-phosphinates (2a–e) prepared, the isopentyl ester (2f) was new.
Then, the alkyl phenyl-H-phosphinates 2a–f were tested in reaction with bromobenzene at 150 °C using 5 mol% Pd(OAc)2 as the catalyst and triethylamine as the base under solvent-free conditions (Scheme 3 and Table 3).
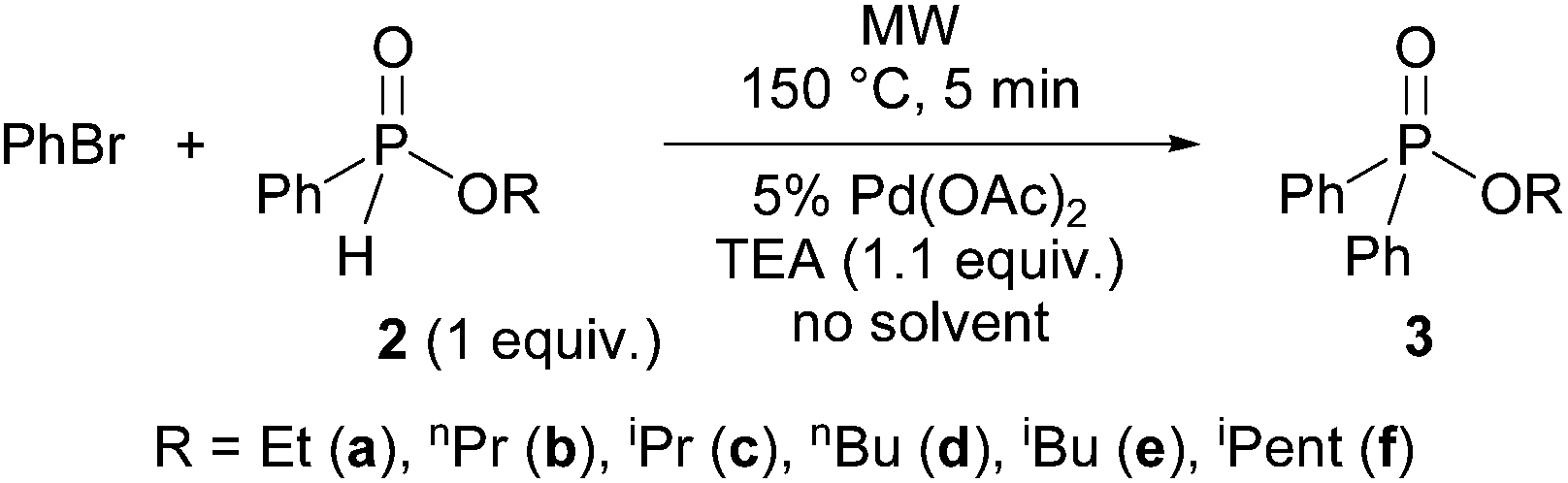 | ||
| Scheme 3 P–C coupling reaction of alkyl phenyl-H-phosphinates (2a–f) with bromobenzene. | ||
| Entry | R | Yield (%) |
|---|---|---|
| 1 | Et | 85 (3a) |
| 2 | nPr | 91 (3b) |
| 3 | iPr | 68 (3c) |
| 4 | nBu | 87 (3d) |
| 5 | iBu | 76 (3e) |
| 6 | iPent | 92 (3f) |
The P–C couplings were complete after a 5 min reaction time. Flash chromatography afforded alkyl diphenylphosphinates 3a, 3b and 3d–f in yields of 76–92% (Table 3, entries 1, 2, 4–6). The sterically hindered isopropyl derivative (3c) was obtained only in a yield of 68% (Table 3, entry 3).
It can be seen, that the alkyl phenyl-H-phosphinates (with the exception of the isopropyl derivative 3c) were as reactive, as dialkyl phosphites in the Pd(OAc)2-catalyzed, P-ligand-free Hirao reaction with bromobenzene. Hence, the P-ligand-free coupling seems to be rather general.
Most of the alkyl diphenylphosphinates (3a, 3c, 3d and 3f) prepared have been described in the literature.1,42–47 All phosphinates (3a–f) were identified by 31P, 13C, 1H NMR, and HR-MS.
As another extension, the coupling of diphenylphosphine oxide with bromobenzene and a few 4-substituted derivatives was also investigated under the standard conditions applied above (Scheme 4). Due to the enhanced reactivity of Ph2P(O)H, the reactions were complete at 120 °C after 15 min, or, using bromobenzene, at 150 °C within 5 min, to afford aryldiphenylphosphine oxides (4a–d) in yields of 83–90% (Table 4). All aryldiphenylphosphine oxides (4a–d) have been known from the literature.48–51
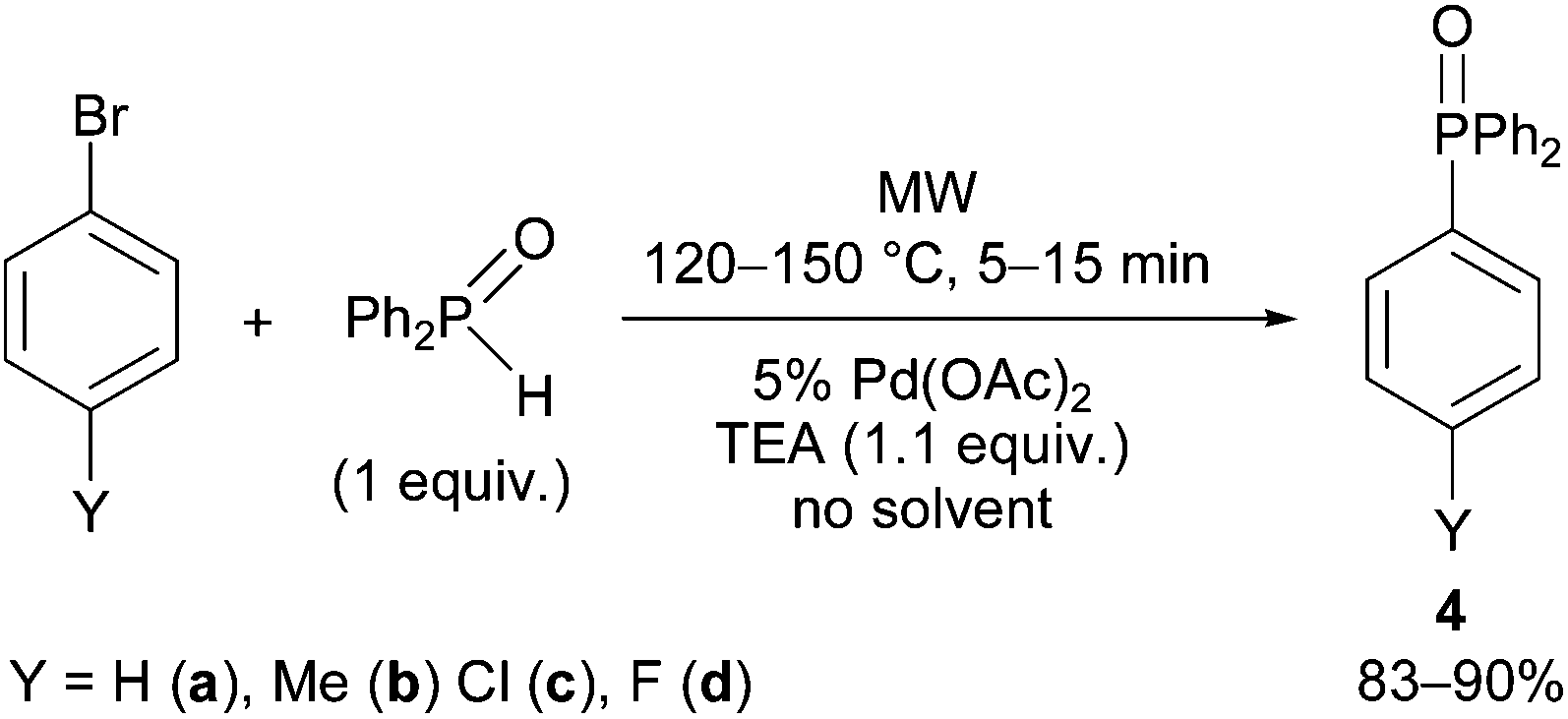 | ||
| Scheme 4 P–C coupling reaction of diphenylphosphine oxide with bromoarenes. | ||
| Entry | Y | T (°C) | t (min) | Yield (%) |
|---|---|---|---|---|
| 1 | H | 120 | 15 | 90 (4a) |
| 2 | H | 150 | <5 | 88 (4a) |
| 3 | 4-Me | 150 | 5 | 83 (4b) |
| 4 | 4-Cl | 150 | 5 | 89 (4c) |
| 5 | 4-F | 150 | 5 | 87 (4d) |
Finally, a few dialkylphenylphosphine oxides (6a–c) were synthesized by the reaction of dialkylphosphine oxides (5a–c) with bromobenzene under the conditions of the P-ligand-free Hirao reaction at 175 °C (Scheme 5). It was advantageous to use acetonitrile as the solvent to avoid the decomposition observed under solvent-free conditions. Using acetonitrile, completion of the reactions required 1.5 h.
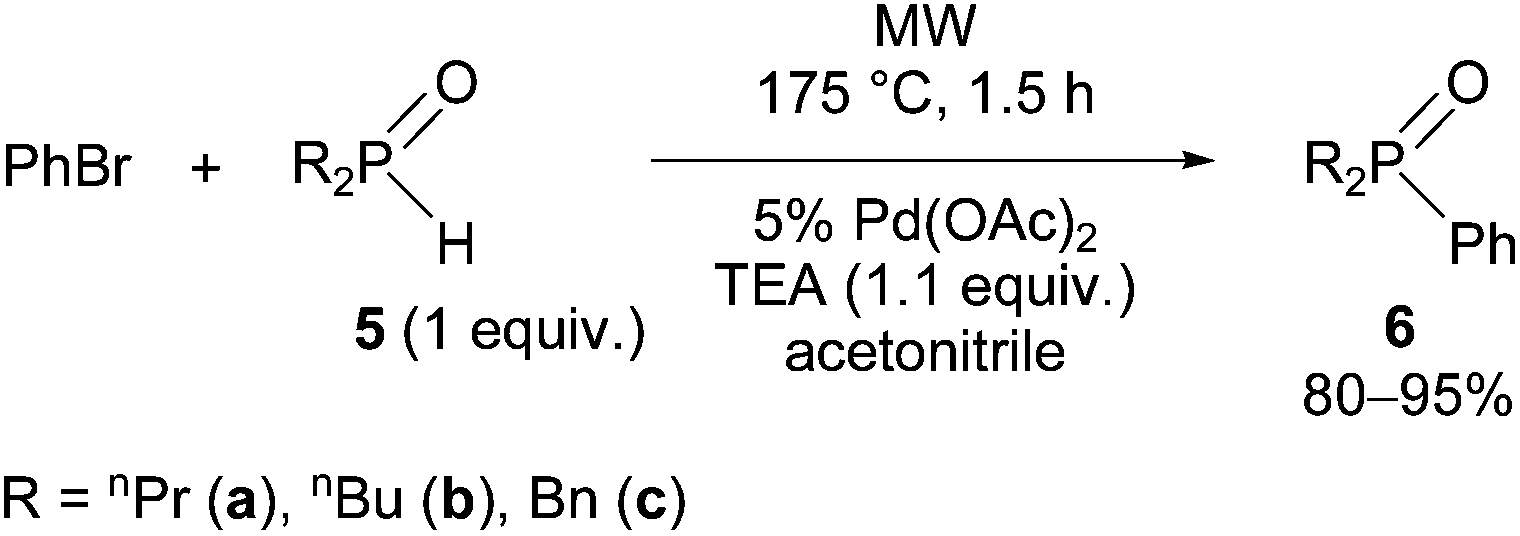 | ||
| Scheme 5 P–C coupling of dialkylphosphine oxides (5a–c) with bromobenzene. | ||
This series of couplings proves the more general value of the P-ligand-free P–C coupling reaction developed by us.
It is noted that the starting secondary phosphines oxides (5a–c)52–55 were also prepared by us.
The dialkylphenylphosphine oxides (6a–c)56–59 prepared are known compounds.
The most striking feature of the newer developments of our work is the broad scope of the possible applications of the new method, since an impressive range of >P(O)H reagents could be used with similar efficiencies. It can be said that the hydrogen atom on the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of diphenylphosphine oxide is significantly more acidic, than the similar proton in diethyl phosphite, as suggested by the pKa values of 14.5 (predicted by the program Marvin Sketch, Version 5.4.1.1) and 20.8,60 respectively. At the same time, the acidity of the P–H of dialkylphosphine oxides is comparable with that of diethyl phosphite or in general dialkyl phosphites. However, the acidity of the P–H has not much role in the efficiency of the couplings studied.
O moiety of diphenylphosphine oxide is significantly more acidic, than the similar proton in diethyl phosphite, as suggested by the pKa values of 14.5 (predicted by the program Marvin Sketch, Version 5.4.1.1) and 20.8,60 respectively. At the same time, the acidity of the P–H of dialkylphosphine oxides is comparable with that of diethyl phosphite or in general dialkyl phosphites. However, the acidity of the P–H has not much role in the efficiency of the couplings studied.
In conclusion, the first P-ligand-free accomplishment of the Hirao reaction has been developed and extended to the synthesis of a wider range of derivatives, such as diethyl arylphosphonates, alkyl diphenylphosphinates and different tertiary phosphine oxides demonstrating the general value of the MW-assisted, P-ligand-free and, in almost all cases, solvent-free conditions for this reaction. The omission of the P-ligand in the Pd-catalyzed Hirao coupling under MW conditions means an enormous advantage from the point of view environmentally friendly conditions and costs. It can be expected that this novel method will have further impact on the development of P-ligand-free methodologies including also other model compounds, such as, among others, (2-bromovinyl)benzenes.
Experimental
General
The reactions were carried out in a 300 W CEM Discover focused microwave reactor equipped with a pressure controller applying 30–50 W under isothermal conditions. Standard 5 mL glass reaction vessels were used distributed by the supplier of the CEM reactor, and the reaction mixtures were stirred magnetically.The 13C and 1H NMR spectra were obtained in CDCl3 solution on a Bruker DRX-500 spectrometer operating at 125.7, and 500.1 MHz, respectively. The 13C and 1H chemical shifts are referred to TMS. 31P NMR spectra were obtained on a Bruker AV-300 spectrometer. Chemical shifts are downfield relative to 85% H3PO4. Mass spectrometry was performed on a ZAB-2SEQ instrument.
1.1. General procedure for the preparation alkyl-phenyl-H-phosphinates. To phenyl-H-phosphinic acid (0.28 g, 2.0 mmol) was added the alkyl bromide (2.4 mmol: 0.19 mL of ethyl iodide, 0.22 mL of propyl bromide, 0.23 mL of isopropyl bromide, 0.26 mL of butyl bromide, 0.26 mL of isobutyl bromide or 0.29 mL of isopentyl bromide) and TEA (0.31 mL, 2.2 mmol) and the resulting mixture was irradiated in a closed vial in the microwave reactor at 80 °C for the time shown in Table 2. The reaction mixture was passed through a thin (ca. 1–1.5 cm) layer of silica gel using ethyl acetate as the eluent. The products (2a–f) were obtained as colourless oils.
Selected spectral data for alkyl phenyl-H-phosphinates
| Entry | Product | δ P (CDCl3) (ppm) | δ (ref. 46) (ppm) | [M + H]+found | [M + H]+requires |
|---|---|---|---|---|---|
| 1 | 2a | 22.7 | 24.7 | 171.0575 | 171.0575 |
| 2 | 2b | 25.7 | 24.9 | 185.0733 | 185.0731 |
| 3 | 2c | 20.6 | 22.3 | 185.0733 | 185.0731 |
| 4 | 2d | 25.1 | 24.9 | 199.0888 | 199.0888 |
| 5 | 2e | 25.7 | 25.0 | 199.0887 | 199.0888 |
Isopentyl phenyl-H-phosphinate (2f). 31P NMR (CDCl3) δ 24.9; 13C NMR (CDCl3) δ 22.4 (CH3), 24.6 (CH), 39.2 (d, J = 6.4, OCH2CH2), 64.5 (d, J = 6.6, OCH2), 128.8 (d, J = 13.8, C2)*, 130.0 (d, J = 132.1, C1), 130.9 (d, J = 11.8, C3)*, 133.1 (d, J = 2.9, C4), * may be reversed; 1H NMR (CDCl3) δ 0.87 (t, 6H, J = 6.1, 2 × CH3), 1.50–1.61 (m, 2H, OCH2CH2), 1.62–1.79 (m, 1H, CH), 4.00–4.15 (m, 2H, OCH2), 7.54 (d, 1H, J = 562.5, P–H), 7.41–7.61 (m, 3H, ArH), 7.69–7.80 (m, 2H, ArH); [M + H]+ = 213.1042, C11H18O2P requires 213.1044.
1.2. General procedure for the preparation of the dialkylphosphine oxides (5a–c). The Grignard reagent (40.0 mmol) formed from (0.97 g, 40.0 mmol) of magnesium and alkyl halide (40.0 mmol: 3.6 mL of bromopropane, 4.2 mL chlorobutane or 4.6 mL of benzyl chloride) in diethyl ether (50 mL) was added dropwise to the diethyl phosphite (1.7 mL, 13.0 mmol) in diethyl ether (10 mL) at 0 °C. The resulting mixture was stirred at 26 °C for 1.5 hours. The mixture was hydrolyzed with 10% HCl solution (40 mL) and the aqueous phase was extracted with diethyl ether (2 × 50 mL). The combined organic phases were dried (Na2SO4). Evaporation of the solvent provided a residue that was purified by column chromatography using silica gel, 1% methanol in dichloromethane as the eluent to give products 5a–c as white crystals.
Dipropylphosphine oxide (5a). Yield: 71%; white crystals; mp.: 47–48 °C, mp.:52 48–50 °C; 31P NMR (CDCl3) δ 35.3, δP (CDCl3)53 32.5; [M + H]+ = 135.0930, C6H16OP requires 135.0933.
Dibutylphosphine oxide (5b). Yield: 65%; white crystals; mp.: 55–56 °C, mp.:54 55–56 °C; 31P NMR (CDCl3) δ 33.9, δP (CDCl3)54 36.09; [M + H]+ = 163.1249, C8H20OP requires 163.1246.
Dibenzylphosphine oxide (5c). Yield: 78%; white crystals; mp.: 109–110 °C, mp.:55 106–107 °C; 31P NMR (CDCl3) δ 36.2, δP (CDCl3)55 35.5; [M + H]+ = 231.0937, C14H16OP requires 231.0933.
2.1. General procedure for the reaction of bromoarenes and diethyl phosphite. To the bromoarene (2.0 mmol: 0.21 mL of bromobenzene, 0.25 mL of 4-bromoanisole, 0.25 mL of 3-bromoanisole, 0.31 mL of 4-propyl bromobenzene, 0.28 mL of 4-ethyl bromobenzene, 0.34 g of 4-bromotoluene, 0.38 g of 4-bromo-chlorobenzene, 0.24 mL of 3-bromo-chlorobenzene, 0.22 mL of 4-bromo-fluorobenzene, 0.22 mL of 3-bromo-fluorobenzene, 0.32 mL of ethyl 4-bromobenzoate, 0.32 mL of ethyl 3-bromobenzoate, 0.40 g of 4-bromoacetophenone and 0.26 mL of 3-bromoacetophenone) was added diethyl phosphite 0.39 mL (3.0 mmol) [or in the case of bromoanisoles, diethyl phosphite (6.0 mmol, 0.79 mL)] triethylamine (0.31 mL, 2.2 mmol) and palladium acetate (0.022 g, 0.10 mmol or 0.044 g, 0.20 mmol – see Table 1.) and the resulting mixture was irradiated in a closed vial in the microwave reactor at the temperature and for the time shown in Table 1. The reaction mixture was passed through a thin (ca. 1.5–2 cm) layer of silica gel using ethyl acetate as the eluent. The products 1a–n were obtained as colourless oils.
Diethyl 4-methoxyphenylphosphonate (1b). 31P NMR (CDCl3) δ 19.8, δP (CDCl3)34 18.8; 13C NMR (CDCl3) δ 16.3 (d, J = 6.6, CH2CH3), 55.3 (OCH3), 62.9 (d, J = 5.3, OCH2), 114.0 (d, J = 16.0, C2)*, 119.6 (d, J = 194.9, C1), 133.8 (d, J = 11.4, C3)*, 162.8 (d, J = 3.3, C4), * may be reversed; 1H NMR (CDCl3) δ 1.31 (t, 6H, J = 7.0, CH3), 3.83 (OCH3), 3.96–4.21 (m, 4H, OCH2), 6.89–7.02 (m, 2H, ArH), 7.65–7.85 (m, 2H, ArH); [M + H]+ = 245.0945, C11H18O4P requires 245.0943.
Diethyl 3-methoxyphenylphosphonate (1c). 31P NMR (CDCl3) δ 18.8; δP (CDCl3)35 18.7; 13C NMR (CDCl3) δ 16.3 (d, J = 6.5, CH2CH3), 55.4 (OCH3), 62.2 (d, J = 5.4, OCH2), 116.4 (d, J = 11.4, C2), 118.8 (d, J = 3.2, C4), 124.0 (d, J = 9.2, C6), 129.6 (d, J = 186.8, C1), 129.8 (d, J = 17.6, C5), 159.5 (d, J = 18.9, C3); 1H NMR (CDCl3) δ 1.30 (t, 6H, J = 7.1, CH3), 3.82 (OCH3), 3.96–4.20 (m, 4H, OCH2), 7.00–7.10 (m, 1H, ArH), 7.26–7.41 (m, 3H, ArH); [M + H]+ = 245.0939, C11H18O4P requires 245.0943.
Diethyl 4-propylphenylphosphonate (1d). 31P NMR (CDCl3) δ 20.5; 13C NMR (CDCl3) δ 13.8 (CH2CH2CH3), 16.4 (d, J = 6.5, CH2CH3), 24.3 (C4–CH2CH2), 38.1 (C4–CH2), 62.0 (d, J = 5.4, OCH2), 125.4 (d, J = 189.9, C1), 128.7 (d, J = 15.4, C2)*, 131.9 (d, J = 10.3, C3)*, 147.6 (d, J = 3.1, C1), * may be reversed; 1H NMR (CDCl3) δ 0.95 (t, 3H, J = 7.3, CH2CH2CH3), 1.32 (t, 6H, J = 7.0, OH2CH3), 1.57–1.75 (m, 2H, C4–CH2CH2), 2.63 (t, 2H, J = 7.6, C4–CH2), 4.00–4.23 (m, 4H, OCH2), 7.23–7.30 (m, 2H, ArH), 7.65–7.78 (m, 2H, ArH); [M + H]+ = 257.1302, C13H22O3P requires 257.1301.
Diethyl 4-ethylphenylphosphonate (1e). 31P NMR (CDCl3) δ 19.6, δP (MeOH)3 19.4; 13C NMR (CDCl3) δ 15.2 (C4–CH2CH3), 16.4 (d, J = 6.5, OCH2CH3), 24.3 (C4–CH2CH3), 61.0 (d, J = 5.3, OCH2), 125.3 (d, J = 190.0, C1), 128.1 (d, J = 15.4, C2)*, 131.9 (d, J = 10.3, C3)*, 149.1 (d, J = 3.1, C4), * may be reversed; 1H NMR (CDCl3) δ 1.24 (t, 3H, J = 7.6, C4–CH2CH3), 1.30 (t, 6H, J = 7.0, OH2CH3), 2.68 (q, 2H, J = 7.6, C4–CH2), 3.97–4.20 (m, 4H, OCH2), 7.21–7.35 (m, 2H, ArH), 7.64–7.80 (m, 2H, ArH); [M + H]+ = 243.1146, C12H20O3P requires 243.1145.
Diethyl 3-chlorophenylphosphonate (1h). 31P NMR (CDCl3) δ 17.5; 13C NMR (CDCl3) δ 16.2 (d, J = 6.4, CH3), 62.3 (d, J = 5.5, CH2), 129.69 (d, J = 9.2, C5), 129.74 (d, J = 16.3, C6), 130.7 (d, J = 187.9, C1), 131.6 (d, J = 10.7, C3), 132.4 (d, J = 3.0, C4), 134.7 (d, J = 20.3, C2); 1H NMR (CDCl3) δ 1.30 (t, 6H, J = 7.1, CH3), 3.96–4.20 (m, 4H, OCH2), 7.40–7.31 (m, 1H, ArH), 7.43–7.51 (m, 1H, ArH), 7.59–7.81 (m, 2H, ArH); [M + H]+ = 249.0448, C10H15O3P35Cl requires 249.0447.
Diethyl 3-fluorophenylphosphonate (1j). 31P NMR (CDCl3) δ 16.7 (d, J = 8.7); δP (CDCl3)37 17.4; 13C NMR (CDCl3) δ 16.3 (d, J1 = 6.4, CH3), 62.4 (d, J1 = 5.5, CH2), 118.6 (dd, J1 = 10.5, J2 = 22.3, C2), 119.5 (dd, J1 = 3.1, J2 = 21.1, C4), 127.5 (dd, J1 = 9.2, J2 = 3.3, C6), 130.5 (dd, J1 = 17.5, J2 = 7.5, C5), 131.0 (dd, J1 = 188.9, J2 = 6.2, C1), 162.4 (dd, J1 = 21.4, J2 = 249.4, C3); 1H NMR (CDCl3) δ 1.29 (t, 6H, J = 7.0, CH3), 3.99–4.20 (m, 4H, OCH2), 7.13–7.23 (m, 1H, ArH), 7.33–7.60 (m, 3H, ArH); [M + H]+ = 233.0743, C10H15O3PF requires 233.0743.
Diethyl 3-ethoxycarbonylphenylphosphonate (1l). 31P NMR (CDCl3) δ 17.4; 13C NMR (CDCl3) δ 14.4 (COCH2CH3), 16.4 (d, J = 6.4, POCH2CH3), 61.4 (COCH2), 62.4 (d, J = 5.5, POCH2), 128.7 (d, J = 15.0, C2), 129.3 (d, J = 189.7, C1), 130.9 (d, J = 15.1, C6), 132.8 (d, J = 10.9, C5), 133.3 (d, J = 3.0, C4), 135.9 (d, J = 10.0, C3), 165.7 (d, J = 2.2, C
O); 1H NMR (CDCl3) δ 1.33 (t, 6H, J = 7.1, POCH2CH3), 1.40 (t, 3H, J = 7.1, COCH2CH3), 4.00–4.28 (m, 4H, POCH2), 4.35–4.46 (m, 2H, COCH2), 7.49–7.60 (m, 1H, ArH), 7.94–8.08 (m, 1H, ArH), 8.22 (d, 1H, J = 7.8, C4–H), 8.46 (d, 1H, J = 13.8, C2–H), [M + H]+ = 287.1048, C13H20O5P requires 287.1048.
Diethyl 3-acetylphenylphosphonate (1n). 31P NMR (CDCl3) δ 18.1, δP (CDCl3)39 18.1; 13C NMR (CDCl3) δ 16.4 (d, J = 6.4, CH2CH3), 26.0 (C(O)CH3), 62.4 (d, J = 5.6, OCH2), 129.0 (d, J = 14.8, C6), 129.5 (d, J = 189.4, C1), 131.7 (d, J = 10.6, C5), 131.9 (d, J = 3.0, C4), 136.0 (d, J = 10.0, C3), 137.2 (d, J = 13.9, C2), 197.2 (d, J = 1.4, C
O); 1H NMR (CDCl3) δ 1.35 (t, 6H, J = 7.0, CH2CH3), 2.65 (3H, C(O)CH3), 4.07–4.28 (m, 4H, OCH2), 7.55–7.66 (m, 1H, ArH), 7.94–8.03 (m, 1H, ArH), 8.15 (d, 1H, J = 7.5, C4–H), 8.38 (d, 1H, J = 13.8, C2–H); [M + H]+ = 257.0943, C12H18O4P requires 257.0943.
2.2. General procedure for the reaction of bromobenzene and alkyl phenyl-H-phosphinates (2a–f). To the bromobenzene (0.11 mL, 1.0 mmol) was added alkyl phenyl-H-phosphinate [1.0 mmol: 0.15 mL of ethyl phenylphosphinate (2a), 0.17 mL of propyl phenylphosphinate (2b), 0.17 mL of isopropyl phenylphosphinate (2c), 0.18 mL of butyl phenylphosphinate (2d), 0.19 mL of isobutyl phenylphosphinate (2e), 0.21 g isopentyl phenylphosphinate (2f)], triethylamine (0.16 mL, 1.1 mmol) and Pd(OAc)2 (0.011 g, 0.05 mmol) and the resulting mixture was irradiated by microwave as above (2.1) at 150 °C for 5 min. The mixture was purified as above using hexane–ethyl acetate 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as the eluent. The products (3a–f) were obtained as white crystals.
:1 as the eluent. The products (3a–f) were obtained as white crystals.
Ethyl diphenylphosphinate (3a). White crystals; mp.: 43–44 °C, mp.:42 39–41 °C, 31P NMR (CDCl3) δ 32.2, δP (CDCl3)43 31.5; 13C NMR (CDCl3) δ 16.5 (d, J = 6.6, CH3), 61.1 (d, J = 5.9, OCH2), 128.4 (d, J = 13.1, C2)*, 131.6 (d, J = 10.1, C3)*, 131.7 (d, J = 137.0, C1), 132.0 (d, J = 2.8, C4), * may be reversed; 1H NMR (CDCl3) δ 1.36 (t, 3H, J = 7.1, CH3), 4.01–4.16 (m, 2H, OCH2), 7.39–7.58 (m, 6H, ArH), 7.75–7.88 (m, 4H, ArH); [M + H]+ = 247.0888, C14H16O2P requires 247.0888.
Propyl diphenylphosphinate (3b). White crystals; mp.: 92–93 °C, mp.:44 89–91 °C; 31P NMR (CDCl3) δ 33.5; 13C NMR (CDCl3) δ 10.3 (CH2CH3), 24.0 (d, J = 6.7, OCH2CH2), 66.4 (d, J = 6.1, OCH2), 128.6 (d, J = 13.1, C2)*, 131.7 (d, J = 10.1, C3)*, 131.8 (d, J = 137.2, C1), 132.2 (d, J = 2.8, C4), * may be reversed; 1H NMR (CDCl3) δ 0.94 (t, 3H, J = 7.4, CH3), 1.71–1.80 (m, 2H, OCH2CH2), 3.99 (q, 2H, J = 6.7, OCH2), 7.39–7.53 (m, 6H, ArH), 7.79–7.88 (m, 4H, ArH); [M + H]+ = 261.1040, C15H18O2P requires 261.1039.
Isopropyl diphenylphosphinate (3c). White crystals; mp.: 101–102 °C, mp.:43 98–100 °C; 31P NMR (CDCl3) δ 30.0, δP (CDCl3)43 31.4; 13C NMR (CDCl3) δ 24.4 (d, J = 4.2, CH3), 70.3 (d, J = 6.0, OCH), 128.5 (d, J = 13.1, C2)*, 131.7 (d, J = 10.1, C3)*, 132.0 (d, J = 2.8, C4), 132.4 (d, J = 137.3, C1), * may be reversed; 1H NMR (CDCl3) δ 1.34 (d, 6H, J = 6.1, CH3), 4.57–4.74 (m, 1H, OCH), 7.36–7.56 (m, 6H, ArH), 7.75–7.90 (m, 4H, ArH); [M + H]+ = 261.1040, C15H18O2P requires 261.1039.
Butyl diphenylphosphinate (3d). White crystals; mp.: 95–96 °C, mp.:45 90–92 °C; 31P NMR (CDCl3) δ 31.2, δP (CDCl3)46 31.2; 13C NMR (CDCl3) δ 13.7 (CH3), 18.9 (CH2CH3), 32.7 (d, J = 6.6, OCH2CH2), 64.8 (d, J = 6.1, OCH), 128.5 (d, J = 13.1, C2)*, 131.7 (d, J = 10.1, C3)*, 131.8 (d, J = 2.8, C4), 132.1 (d, J = 137.0, C1), * may be reversed; 1H NMR (CDCl3) δ 0.92 (t, 3H, J = 7.3, CH3), 1.35–1.55 (d, 2H, CH2CH3), 1.62–1.80 (m, 2H, OCH2CH2), 4.03 (t, 2H, J = 6.6, OCH2), 7.35–7.61 (m, 6H, ArH), 7.71–7.94 (m, 4H, ArH); [M + H]+ = 275.1196, C16H20O2P requires 275.1195.
Isobutyl diphenylphosphinate (3e). White crystals; mp.: 82–83 °C, mp.:44 79–80 °C; 31P NMR (CDCl3) δ 31.0; 13C NMR (CDCl3) δ 19.0 (CH3), 29.4 (d, J = 6.9, CH), 70.9 (d, J = 6.3, OCH2), 128.6 (d, J = 13.1, C2)*, 131.8 (d, J = 10.1, C3)*, 131.9 (d, J = 2.8, C4), 132.2 (d, J = 137.2, C1), * may be reversed; 1H NMR (CDCl3) δ 0.96 (d, 6H, J = 6.7, CH3), 1.89–2.07 (m, 1H, CH), 3.78 (t, 2H, J = 6.3, OCH2), 7.34–7.56 (m, 6H, ArH), 7.65–7.88 (m, 4H, ArH); [M + H]+ = 275.1197, C16H20O2P requires 275.1195.
Isopentyl diphenylphosphinate (3f). White crystals; mp.: 55–56 °C, mp.:47 55–57 °C; 31P NMR (CDCl3) δ 31.2, δP (CDCl3)47 30.1; 13C NMR (CDCl3) δ 22.3 (CH3), 24.6 (CH), 39.2 (d, J = 6.6, OCH2CH2), 63.4 (d, J = 6.1, OCH2), 128.4 (d, J = 13.1, C2)*, 131.5 (d, J = 10.1, C3)*, 131.6 (d, J = 137.0, C1), 132.0 (d, J = 2.8, C4), * may be reversed; 1H NMR (CDCl3) δ 0.88 (d, 6H, J = 6.5, CH3), 1.60 (q, 2H, J = 6.7, OCH2CH2), 1.68–1.85 (m, 1H, CH), 4.04 (q, 2H, J = 6.5, OCH2), 7.34–7.56 (m, 6H, ArH), 7.65–7.85 (m, 4H, ArH); [M + H]+ = 289.1353, C17H22O2P requires 289.1352.
2.3. General procedure for the reaction of secondary phosphine oxides with bromoarenes. The tertiary phosphine oxides (4a–d and 6a–c) were prepared from the corresponding bromoarene (2.0 mmol: 0.21 mL of bromobenzene, 0.34 g of 4-bromotoluene, 0.38 g 4-bromo-chlorobenzene and 0.22 mL 4-bromo-fluorobenzene) and the secondary phosphine oxide [2.0 mmol: 0.40 g of diphenylphosphine oxide, 0.27 g of dipropylphosphine oxide (5a), 0.32 g of dibutylphosphine oxide (5b) and 0.46 g of dibenzylphosphine oxide (5c)] [in the case of dialkylphosphine oxides in acetonitrile (1 mL)], as above (2.1). The only difference is that, in the case of dialkylphosphine oxides (5a–c), the solvent was removed after the reaction. The products 4a–d and 6a–c were obtained as white or pale yellow crystals.
Triphenylphosphine oxide (4a). White crystals; mp.: 156–157 °C, mp.:48 156–157 °C; 31P NMR (CDCl3) δ 30.6, δP (CDCl3)48 29.5. [M + H]+ = 279.0941, C18H16OP requires 279.0939.
4-Methylphenyl diphenylphosphine oxide (4b). White crystals; mp.: 118–119 °C, mp.:49 119–123 °C; 31P NMR (CDCl3) δ 29.3, δP (CDCl3)49 29.1; 13C NMR (CDCl3) δ 21.6 (CH3), 128.4 (d, J = 12.1, C′2)a, 129.1 (d, J = 106.4, C1), 129.2 (d, J = 12.6, C2)b, 131.8 (d, J = 2.7, C′4), 132.0 (d, J = 9.9, C′3)a, 132.1 (d, J = 10.2, C3)b, 132.8 (d, J = 105.9, C′1), 142.4 (d, J = 2.8, C4), a,b may be reversed; 1H NMR (CDCl3) δ 2.40 (s, 3H, CH3), 7.11–7.29 (m, 2H, ArH), 7.40–7.80 (m, 12H, ArH); [M + H]+ = 293.1085, C19H18OP requires 293.1090.
4-Chlorophenyl diphenylphenylphosphine oxide (4c). Pale yellow crystals; mp.: 142–143 °C, mp.:49 141–142 °C; 31P NMR (CDCl3) δ 28.5, δP (CDCl3)49 28.8; 13C NMR (CDCl3) δ 128.6 (d, J = 12.2, C′2)a, 128.8 (d, J = 12.7, C2)b, 131.1 (d, J = 104.7, C1), 131.96 (d, J = 10.0, C′3)a, 132.02 (d, J = 105.0, C′1), 132.1 (d, J = 2.7, C′4), 133.4 (d, J = 10.7, C3)b, 138.5 (d, J = 3.4, C4), a, b may be reversed; 1H NMR (CDCl3) δ 7.20–7.87 (m, ArH); [M + H]+ = 313.0551, C18H15OP35Cl requires 313.0544.
4-Fluorophenyl diphenylphosphine oxide (4d). Pale yellow crystals; mp.: 134–135 °C, mp.:50 134–135 °C; 31P NMR (CDCl3) δ 28.5, δP (CDCl3)51 28.3; 13C NMR (CDCl3) δ, 115.8 (dd, J1 = 13.2, J2 = 21.4, C2)a, 128.52 (dd, J1 = 106.5, J2 = 3.4, C1), 128.53 (d, J = 12.2, C′2)b, 132.0 (d, J = 12.2, C′3)b, ∼132.1 (d, J ∼3.0, C′4), 132.3 (d, J = 105.0, C′1), 134.5 (dd, J1 = 11.3, J2 = 8.8, C3)a, 165.0 (dd, J1 = 3.2, J2 = 253.6, C4), a,b may be reversed; 1H NMR (CDCl3) δ 7.06–7.20 (m, 2H, ArH), 7.37–7.75 (m, 12H, ArH), [M + H]+ = 297.0846, C18H15OPF requires 297.0839.
Dipropylphenylphosphine oxide (6a). Yield: 80%; pale yellow crystals; mp.: 41–42 °C, mp.:56 43 °C; 31P NMR (CDCl3) δ 40.5; 13C NMR (CDCl3) δ 15.4 (d, J = 4.1, CH3), 15.9 (d, J = 14.9, CH2CH3), 32.3 (d, J = 68.3, PCH2), 128.8 (d, J = 11.1, C2)*, 130.6 (d, J = 8.8, C3)*, 131.6 (d, J = 2.7, C4), 132.8 (d, J = 91.7, C1), * may be reversed; 1H NMR (CDCl3) δ 0.95 (t, 6H, J = 7.3, CH3), 1.37–1.69 (m, 4H, CH2CH3), 1.75–2.01 (m, 4H, PCH2), 7.40–7.51 (m, 3H, ArH), 7.61–7.73 (m, 2H, ArH); [M + H]+ = 211.1247, C12H20OP requires 211.1246.
Dibutylphenylphosphine oxide (6b). Yield: 88%; pale yellow crystals; mp.: 58–59 °C, mp.:57 55–57 °C; 31P NMR (CDCl3) δ 39.1, δP (CDCl3)58 42.8; 13C NMR (CDCl3) δ 13.5 (CH3), 23.4 (d, J = 4.1, CH2CH3), 24.0 (d, J = 14.5, PCH2CH2), 29.6 (d, J = 68.5, PCH2), 128.5 (d, J = 11.1, C2)*, 130.3 (d, J = 8.7, C3)*, 131.3 (d, J = 2.7, C4), 132.6 (d, J = 91.8, C1), * may be reversed; 1H NMR (CDCl3) δ 0.83 (t, 6H, J = 6.8, CH3), 1.23–1.69 (m, 8H, CH2CH2), 1.74–2.05 (m, 4H, PCH2), 7.37–7.54 (m, 3H, ArH), 7.59–7.75 (m, 2H, ArH); [M + H]+ = 239.1554, C14H24OP requires 239.1559.
Dibenzylphenylphosphine oxide (6c). Yield: 95%; white crystals; mp.: 179–180 °C, mp.:59 178–181 °C; 31P NMR (CDCl3) δ 35.0, δP (CDCl3)59 35.2; 13C NMR (CDCl3) δ 37.4 (d, J = 63.4, PCH2), 126.7 (d, J = 2.9, C′4), 128.2 (d, J = 11.4, C2)a, 128.4 (d, J = 2.5, C′3)b, 129.9 (d, J = 5.2, C′2)b, 130.9 (d, J = 94.8, C1), 131.0 (d, J = 8.5, C3)a, 131.4 (d, J = 7.6, C′1), 131.6 (d, J = 2.7, C4), a, b may be reversed; 1H NMR (CDCl3) δ 3.49 (d, 4H, J = 13.9, PCH2), 7.20–7.71 (m, 15H, ArH); [M + H]+ = 307.1241, C20H20OP requires 307.1246.
Acknowledgements
The above project was supported by the Hungarian Scientific and Research Fund (OTKA no. K83118).References
- Preceding communication: E. Jablonkai and G. Keglevich, Tetrahedron Lett., 2013, 54, 4185 CrossRef CAS PubMed.
- E. Jablonkai and G. Keglevich, Curr. Org. Synth., 2014, 11, 429 CrossRef CAS.
- C. Yuan and H. Feng, Synthesis, 1990, 140 CrossRef CAS.
- G. Keglevich, A. Grün, A. Bölcskei, L. Drahos, M. Kraszni and G. T. Balogh, Heteroat. Chem., 2012, 23, 574 CrossRef CAS.
- T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Tetrahedron Lett., 1980, 21, 3595 CrossRef CAS.
- T. Hirao, T. Masunaga, N. Yamada, Y. Ohshiro and T. Agawa, Bull. Chem. Soc. Jpn., 1982, 55, 909 CrossRef CAS.
- T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Synthesis, 1981, 56 CrossRef CAS.
- X. Lu and J. Zhu, Synthesis, 1987, 726 CrossRef CAS.
- D. A. Holt and J. M. Erb, Tetrahedron Lett., 1989, 30, 5393 CrossRef CAS.
- M. A. Kazankova, I. G. Trostyanskaya, S. V. Lutsenko and I. P. Beletskaya, Tetrahedron Lett., 1999, 40, 569 CrossRef CAS.
- P. Zhong, Z. X. Xiong and X. Huang, Synth. Commun., 2000, 30, 273 CrossRef CAS.
- Y. Kobayashi and A. D. William, Adv. Synth. Catal., 2004, 346, 1749 CrossRef CAS.
- M. Kalek, A. Ziadi and J. Stawinski, Org. Lett., 2008, 10, 4637 CrossRef CAS PubMed.
- D. Villemin, P.-A. Jaffres and F. Simeon, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 130, 59 CrossRef CAS.
- M. Kalek and J. Stawinski, Organometallics, 2007, 26, 5840 CrossRef CAS.
- Y. Belabassi, S. Alzghari and J.-L. Montchamp, J. Organomet. Chem., 2008, 693, 3171 CrossRef CAS PubMed.
- E. L. Deal, C. Petit and J.-L. Montchamp, Org. Lett., 2011, 13, 3270 CrossRef CAS PubMed.
- M. Kalek, M. Jezowska and J. Stawinski, Adv. Synth. Catal., 2009, 351, 3207 CrossRef CAS.
- M. M. Kabachnik, M. D. Solntseva, V. V. Izmer, Z. S. Novikova and I. P. Beletskaya, Russ. J. Org. Chem., 1998, 34, 93 CAS.
- I. P. Beletskaya, N. B. Karlstedt, E. E. Nifant'ev, D. V. Khodarev, T. S. Kukhareva, A. V. Nikolaev and A. J. Ross, Russ. J. Org. Chem., 2006, 42, 1780 CrossRef CAS.
- Z. S. Novikova, N. N. Demik, A. Yu. Agarkov and I. P. Beletskaya, Russ. J. Org. Chem., 1995, 31, 129 Search PubMed.
- I. P. Beletskaya and M. A. Kazankova, Russ. J. Org. Chem., 2002, 38, 1391 CrossRef CAS.
- A. Stadler and C. O. Kappe, Org. Lett., 2002, 4, 3541 CrossRef CAS PubMed.
- L. Liu, Y. Wang, Z. Zeng, P. Xu, Y. Gao, Y. Yin and Y. Zhao, Adv. Synth. Catal., 2013, 355, 659 CrossRef CAS.
- L. Liu, Y. Lv, Y. Wu, X. Gao, Z. Zeng, Y. Gao, G. Tang and Y. Zhao, RSC Adv., 2014, 4, 2322 RSC.
- G. Keglevich, T. Novák, L. Vida and I. Greiner, Green Chem., 2006, 8, 1073 RSC.
- G. Keglevich, K. Majrik, L. Vida and I. Greiner, Lett. Org. Chem., 2008, 5, 224 CrossRef CAS.
- I. Greiner, A. Grün, K. Ludányi and G. Keglevich, Heteroat. Chem., 2011, 22, 11 CrossRef CAS.
- G. Keglevich, A. Grün, Z. Blastik and I. Greiner, Heteroat. Chem., 2011, 22, 174 CrossRef CAS.
- A. Grün, Z. Blastik, L. Drahos and G. Keglevich, Heteroat. Chem., 2012, 23, 241 CrossRef.
- G. Keglevich, E. Bálint, É. Karsai, A. Grün, M. Bálint and I. Greiner, Tetrahedron Lett., 2008, 49, 5039 CrossRef CAS PubMed.
- G. Keglevich, E. Bálint, É. Karsai, J. Varga, A. Grün, M. Bálint and I. Greiner, Lett. Org. Chem., 2009, 6, 535 CrossRef CAS.
- K. Kranjc and M. Kočevar, Curr. Org. Chem., 2010, 14, 1050 CrossRef CAS.
- M. C. Kohler, J. G. Sokol and R. A. Stockland Jr, Tetrahedron Lett., 2009, 50, 457 CrossRef CAS PubMed.
- R. A. Dhokale and S. B. Mhaske, Org. Lett., 2013, 15, 2218 CrossRef CAS PubMed.
- R. Q. Zhuang, J. A. Xu, Z. S. Cai, G. Tang, M. J. Fang and Y. F. Zhao, Org. Lett., 2011, 13, 2110 CrossRef CAS PubMed.
- P. Machnitzki, T. Nickel, O. Stelzer and C. Landgrafe, Eur. J. Inorg. Chem., 1998, 7, 1029 CrossRef.
- C. A. Metcalf, W. C. Shakespeare, T. K. Sawyer, Y. Wang and R. Bohacek, US Pat., 2003/100572 A1, 2003.
- E. A. Krasil'nikova, I. V. Berdnik, V. V. Sentemov, F. S. Shagvaleev and T. V. Zykova, J. Gen. Chem. USSR, 1986, 56, 959 Search PubMed.
- E. Bálint, E. Jablonkai, M. Bálint and G. Keglevich, Heteroat. Chem., 2010, 21, 211 Search PubMed.
- G. Keglevich, E. Bálint, N. Z. Kiss, E. Jablonkai, L. Hegedűs, A. Grün and I. Greiner, Curr. Org. Chem., 2011, 15, 1802 CrossRef CAS.
- A. W. Frank and C. F. Baranauckas, J. Org. Chem., 1966, 31, 872 CrossRef CAS.
- T. K. Olszewski and B. Boduszek, Tetrahedron, 2010, 66, 8661 CrossRef CAS PubMed.
- K. D. Berlin and R. U. Pagilagan, J. Org. Chem., 1967, 32, 129 CrossRef CAS.
- O. Berger, C. Petit, E. L. Deal and J.-L. Montchamp, Adv. Synth. Catal., 2013, 355, 1361 CrossRef CAS.
- N. Z. Kiss, K. Ludányi, L. Drahos and G. Keglevich, Synth. Commun., 2009, 39, 2392 CrossRef CAS.
- D. B. G. Williams and T. E. Netshiozwi, Tetrahedron, 2009, 65, 9973 CrossRef CAS PubMed.
- X. Zhang, H. Liu, X. Hu, G. Tang, J. Zhu and Y. Zhao, Org. Lett., 2011, 13, 3478 CrossRef CAS PubMed.
- C. Huang, X. Tang, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2006, 71, 5020 CrossRef CAS PubMed.
- J. W. Rakshys, R. W. Taft and W. A. Sheppard, J. Am. Chem. Soc., 1968, 90, 5236 CrossRef.
- J. Xu, P. Zhang, Y. Gao, Y. Chen, G. Tang and Y. Zhao, J. Org. Chem., 2013, 78, 8176 CrossRef CAS PubMed.
- M. I. Kabachnik and E. N. Tsvetkov, Bull. Acad. Sci. USSR, 1963, 12, 1120 CrossRef.
- S. F. Malysheva, Z. M. Garashchenko, S. N. Arbuzova, M. V. Nikitin, N. K. Gusarova and B. A. Trofimov, Russ. J. Gen. Chem., 1997, 67, 1794 CAS.
- C. A. Busacca, J. C. Lorenz, N. Grinberg, N. Haddad, M. Hrapchak, B. Latli, H. Lee, P. Sabila, A. Saha, M. Sarvestani, S. Shen, R. Varsolona, X. Wei and C. H. Senanayake, Org. Lett., 2005, 7, 4277 CrossRef CAS PubMed.
- B. A. Trofimov, N. K. Gusarova, S. F. Malysheva, S. I. Shaikhudinova, N. A. Belogorlova, T. I. Kazantseva, B. G. Sukhov and G. V. Plotnikova, Russ. J. Gen. Chem., 2005, 75, 684 CrossRef CAS PubMed.
- J. H. Davies and P. Kirby, J. Chem. Soc., 1964, 3425 RSC.
- V. I. Evreinov, V. E. Baulin, Z. N. Vostroknutova, Z. V. Safronova, N. A. Bondarenko and E. N. Tsvetkov, Russ. J. Gen. Chem., 1995, 65, 223 Search PubMed.
- V. A. Zagumennov and N. A. Sizova, Russ. J. Gen. Chem., 2012, 82, 1368 CrossRef CAS.
- M. Stankevič, A. Włodarczyk, M. Jaklińska, R. Parcheta and K. M. Pietrusiewicz, Tetrahedron, 2011, 67, 8671 CrossRef PubMed.
- K. D. Troev, Chemistry and Application of H-Phosphonates, Elsevier, Amsterdam, 2006, ch. 3.1, p. 24 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03292f |
| This journal is © The Royal Society of Chemistry 2014 |