
Review on modified N–TiO2 for green energy applications under UV/visible light: selected results and reaction mechanisms
Abstract
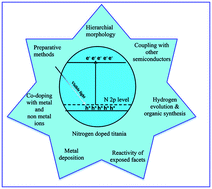
Titanium dioxide photocatalyst has witnessed considerable interest for use in water and air cleanup owing to its fascinating properties like non-toxicity, ease of preparation, favorable band edge positions, water insolubility, multifaceted electronic properties, surface acid–base properties, and super hydrophilicity. Although it possesses much functionality, large bandgap and massive charge carrier recombination limits its wide utility under natural solar light. These drawbacks were overcome through nitrogen doping in titania matrix (N–TiO2), which alters the surface-bulk structure for visible light absorption with high quantum efficiency. In this review, we highlight the recent progress of N–TiO2 towards pollutant degradation, hydrogen evolution and its use in organic synthesis under ambient conditions. The preparation of N–TiO2 via different methods (physical and chemical methods) with diverse morphologies, nature of chemical dopants, induced defects and fundamental reaction parameters governing efficient photoinduced reactions are explored in this review. Further improvements in the photoefficiency of N–TiO2 were achieved through co-doping with foreign ions, heterostructuring with other semiconductors, metal deposition and the tuning of N–TiO2 with reactive exposed facets. The resultant improvement from each of the modification is discussed in the light of charge carrier generation–separation–transfer–recombination dynamics together with pollutant adsorption and their reactions with reactive oxygenated species in the liquid or gaseous regime. This review attempts to give an overview of the research highlights concerned with N–TiO2. Although it is impossible to cover all of the research articles in the literature, several milestones in the pathway towards highly applicable N–TiO2 are explored. It is hoped that this review article will trigger further research in synthesizing N–TiO2 with multifunctional features to enhance its capacity for green energy applications.
 Please wait while we load your content...
Please wait while we load your content...

