Adsorption, photodegradation and antibacterial study of graphene–Fe3O4 nanocomposite for multipurpose water purification application†
Chella Santhosha,
Pratap Kollu‡*b,
Sejal Doshic,
Madhulika Sharmac,
Dhirendra Bahadur*c,
Mudaliar T. Vanchinathana,
P. Saravanand,
Byeong-Su Kime and
Andrews Nirmala Grace*a
aCentre for Nanotechnology Research, VIT University, Vellore 632014, India. E-mail: anirmalagladys@gmail.com; anirmalagrace@vit.ac.in; Tel: +919791322311
bDST-INSPIRE Faculty, Department of Metallurgical Engineering and Materials Science, Indian Institute of Technology Bombay, Mumbai 400076, India
cDepartment of Metallurgical Engineering and Materials Science, Indian Institute of Technology Bombay, Mumbai 400076, India. E-mail: dhirenb@iitb.ac.in
dDefence Metallurgical Research Laboratory, Hyderabad 500 058, India
eDepartment of Chemistry and Department of Energy Engineering, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan 689-798, Korea
First published on 3rd June 2014
Abstract
Graphene–Fe3O4 (G–Fe3O4) composite was prepared from graphene oxide (GO) and FeCl3·6H2O by a one-step solvothermal route. The as-prepared composite was characterized by field-emission scanning electron microscopy, transmission electron microscopy, dynamic light scattering and X-ray powder diffraction. SEM analysis shows the presence of Fe3O4 spheres with size ranging between 200 and 250 nm, which are distributed and firmly anchored onto the wrinkled graphene layers with a high density. The resulting G–Fe3O4 composite shows extraordinary adsorption capacity and fast adsorption rates for the removal of Pb metal ions and organic dyes from aqueous solution. The adsorption isotherm and thermodynamics were investigated in detail, and the results show that the adsorption data was best fitted with the Langmuir adsorption isotherm model. From the thermodynamics investigation, it was found that the adsorption process is spontaneous and endothermic in nature. Thus, the as-prepared composite can be effectively utilized for the removal of various heavy metal ions and organic dyes. Simultaneously, the photodegradation of methylene blue was studied, and the recycling degradation capacity of dye by G–Fe3O4 was analyzed up to 5 cycles, which remained consistent up to ∼97% degradation of the methylene blue dye. Although iron oxide has an affinity towards bacterial cells, its composite with graphene still show antibacterial property. Almost 99.56% cells were viable when treated with Fe3O4 nanoparticle, whereas with the composite barely 3% cells survived. Later, the release of ROS was also investigated by membrane and oxidative stress assay. Total protein degradation was analyzed to confirm the effect of the G–Fe3O4 composite on E. coli cells.
Introduction
Water pollution resulting from biotic and abiotic pollutants has been a major environmental threat attributable to both existing and emerging industries. Heavy metals and dyes beyond the permissible limits directly cause toxicity to humans and other living organisms. Metal ions, such as Cd(II) and Pb(II) have been reported to immobilize the enzymes in some plants and can also cause energy deficiency in the transmission of nerve impulses in humans.1,2 Hence, it has become immediately necessary to prevent water pollution caused by microbes as well as by heavy metals and dyes. Most purification methods suffer from some drawbacks such as high capital and operational costs or disposal of residual metal sludge, and hence are not suitable for small scale industries.3 In addition, these methods are not very efficient at low concentrations in the range 1–100 mg L−1.4,5 Silver has become the basis for antibacterial activity; however, because silver is expensive and leaches into the environment, the focus has shifted to the exploration of inexpensive materials for antibacterial activity.Adsorption is one of the promising processes for the removal of heavy metal ions from water. Some of the adsorbent materials for heavy metal ion removal are activated carbon (powder or granular) and CNTs, etc.,6–8 for the removal of Pb, cadmium and other heavy metal ions. However, the cost of adsorbent becomes relatively high when pure sorbents are used. Photodegradation of dye has been a successful method for the eradication of dyes from effluent water, which further helps in water purification.
Graphene and Fe3O4 (ref. 9 and 10) individually have been reported for their filtration and purification activity. For standalone use they can be used as water-purifying materials, thus it was considered to combine them as a composite and efficiently use them as a nanomaterial for water purification. Graphene is a single layer of carbon densely packed in a honeycomb crystal lattice structure. In comparison with other carbonaceous material, it has drawn much attention since its discovery due to its unique electronic and mechanical properties.11,12 Its remarkable properties, such as large surface area (2630 m2 g−1), good chemical stability, flat structure, imply that it can be used as an excellent adsorbent.13–16 Its basal plane structure allows for strong π–π interactions, assigned to the π–π stacking between aromatic dyes and the π-conjugation regions of the graphene layers.17This property of graphene would serve as a good adsorbent for removing pollutants such as metal ions, dyes, and biological materials. However, graphene sheets usually suffer from serious agglomeration and restacking due to their π–π interactions between neighbouring sheets, leading to a significant loss of effective surface area and lower adsorption capacity.18,19 To overcome such difficulties, magnetic adsorbents have emerged as new generation materials for environmental application. The advantage of such materials is their ability to effect magnetic separation by applying an external magnetic field to extract the adsorbent material from the suspension.20,21
Compared with traditional methods, such as filtration, centrifugation and gravitational separation, magnetic separation requires less energy and better separation can be achieved. Therefore, the synthesis of graphene with covalently attached Fe3O4 offers an effective approach to overcome the separation problem associated with graphene. At the same time, the attachment of Fe3O4 with graphene provides a decrease in the possibility of serious agglomeration. In this regard, the current work is focused on the synthesis of G–Fe3O4 composites by a solvothermal route. Further, the as-prepared composites were tested for adsorption properties towards Pb ions and the photodegradation of methylene blue. To better understand its antimicrobial mechanism, we compared the antibacterial activity of graphene and G–Fe3O4 towards a bacterial model, Escherichia coli. Under similar concentration and incubation conditions, G–Fe3O4 dispersion shows the highest antibacterial activity. ROS22 and protein degradation were responsible for bacterial cell wall rupture.
Experimental
Materials
All chemicals were analytical grade, purchased from Sigma Aldrich and used as received without purification. Graphite powder, hydrogen peroxide (30 wt%), sodium nitrate (98%), sulphuric acid (98 wt%), potassium permanganate, Pb(NO3)2, FeCl3·6H2O and sodium acetate (NaAc) were purchased from SD-Fine and used as received.Preparation of graphene oxide
Graphene oxide (GO) was prepared from natural graphite powder by the modified Hummers method.23 In a typical synthesis, 1 g of graphite was added to 23 mL of 98% H2SO4, followed by stirring at room temperature over a period of 24 h. Then, 100 mg of NaNO3 was added to the mixture, stirred for 30 min and then maintained below 5 °C in an ice bath. Finally, 3 g of KMnO4 was added slowly into the mixture. Afterwards, the mixture was heated to 35–40 °C with continuous stirring for 30 min. Then, 46 mL of water was added to the abovementioned mixture and mixed well for 25 min. Finally, 140 mL of water and 10 mL of 30% H2O2 were added to the mixture to stop the reaction. Unreacted graphite in the resulting mixture was removed by centrifugation and the resulting mixture was dried at 60 °C in a vacuum oven. The as-synthesized GO was dispersed into individual sheets in distilled water at a concentration of 0.5 mg mL−1 with the help of ultrasound for further use.Synthesis of G-Fe3O4nanocomposites
The as-prepared GO was used as a precursor for the preparation of G–Fe3O4 composites by a one-step solvothermal method. In a typical synthesis, the as-prepared GO (0.5 g) was exfoliated by ultrasonication in 80 mL of ethylene glycol for more than 3 h. 1.6 g FeCl3·6H2O and 3.2 g NaAc were then dissolved in GO/EG solution at ambient temperature. After stirring for about 30 min, the solution was transferred to a 100 mL teflon-lined stainless-steel autoclave and maintained at 200 °C for 6 h, followed by naturally cooling to ambient temperature. The black precipitate was centrifuged, washed with ethanol several times and finally dried at 60 °C in a vacuum oven.Characterization
The particle size and morphology of the material was studied by FE-Scanning Electron Microscopy (FE-SEM) (HITACHI SU6600 SEM). The phase and crystallographic structure were identified by X-ray diffraction (XRD; Philips X′Pert Pro, Cu-Kα: λ = 0.1540598 nm). The adsorbed metal ion concentration was examined by an atomic adsorption spectrophotometer (Perkin-Elmer Analyst 240). Thermogravimetric analysis (TGA) of the samples was performed on an SDT Q600 (TA Instruments, Korea) with a heating rate of 5 °C min−1 from 0° to 1000 °C. The G–Fe3O4 samples were characterized with a high resolution transmission electron microscope (HR-TEM) JEOL-2000EX operated at 120 kV. Dynamic light scattering measurements were performed using a Beckman Coulter Delsa™ Nano instrument.Analytical measurement
For analytical studies, analytical grade lead nitrate was used to prepare a stock solution containing 1000 mg L−1 of Pb(NO3)2, which was further diluted with double-distilled deionized water to the required ionic concentration. Adsorption thermodynamic experiments were conducted in a 100 mL glass flask containing 25 mg of adsorbent and 100 mL of Pb ion solution at varying concentrations (10–50 mg L−1) and pH 5. The samples were then placed in an orbital shaker for continuous stirring at 250 rpm for different durations and at selected temperatures (300, 310, and 320 K). After a certain time, the suspension was filtered using 0.22 μm cellulose nitrate membrane, and the filtrates were immediately examined using atomic adsorption spectrophotometry (AAS) to measure the ion concentration. The difference between the initial and the equilibrium ion concentration gave the amount of ion adsorbed to the G–Fe3O4 surfaces.Batch mode adsorption
The effects of experimental parameters, such as initial concentration (10–50 mg L−1), pH (3–8), and temperature, were studied in a batch mode of adsorption for specific contact times (0–180 min). To determine the effect of each parameter, the other parameters were maintained constant. Pb solution was prepared by dissolving Pb(NO3)2 in double distilled water and used as the stock solution. Then, the solutions were diluted to the required concentration for the experimental study. The pH of the solution was adjusted using 0.1 M HCl or 0.1 M NaOH. For contact time measurements, 100 mL of Pb ion solution containing 10 mg L−1 of an initial ion concentration at pH 5 was put into a 250 mL conical flask with a fixed amount of adsorbent (25 mg L−1) and agitated in an orbital shaker at 250 rpm and 310 K. At various intervals, the adsorbent were separated from the samples by filtering, and the filtrates were analyzed by AAS to determine the concentration of each ion in the solution.24 The adsorption percentage of a metal ion was calculated as follows:
 | (1) |
Photodegradation activity of G-Fe3O4
The photodegradation activity of the as-prepared G–Fe3O4 was evaluated by the photodegradation of methylene blue dye using 60 W of a visible light CFL lamp. For the degradation of dye, 0.2 g L−1 of photocatalyst was added to 100 mL of methylene blue dye (10 mg L−1) aqueous solution. 1 mL of 30% H2O2 was added to the reaction mixture at the beginning of light irradiation. About 1 mL of aliquot was withdrawn at a given irradiation time and then magnetically separated to remove the catalyst. In the case of bare graphene nanosheets, the suspension was separated by centrifuging at 3000 rpm. The concentration of the remaining dye was determined by measuring the absorbance of solutions at 664 nm by UV-vis spectroscopy.Biocompatibility of G-Fe3O4
Sulforhodamine-B (SRB) assay was performed to evaluate the biocompatibility of G–Fe3O4 with normal mouse fibroblast cells (L929). Although iron oxide nanoparticles have been well-established as biocompatible nanomaterials, it is essential to evaluate the toxic effect of G–Fe3O4 on cell proliferation and morphology of L929 cells because of the presence of graphene. It is important to test the toxic effect of functional material towards a living system. The cells were seeded into 96-well plates at a density of 1 × 104 cells per well and incubated for 24 h in a 5% CO2 environment. Then, 200 μL of different concentrations of the dispersed suspension of G–Fe3O4 (2.0, 1.0, 0.5, 0.25, 0.125, 0.625, and 0.3125 mg mL−1) in DMEM growth medium were added to the cells and incubated for another 24 h at 37 °C and 5% CO2 environment. Thereafter, the cells were gently washed with phosphate buffer saline (PBS; pH 7.3) and processed for SRB assay to determine the viable cell population. Non-treated cells were used as control for the experiments. The cells were then fixed with 10% trichloroacetic acid solution by incubating at 4 °C for 1 h. This was followed by gentle washing of cells with water and then staining with 0.4% SRB dissolved in 1% acetic acid and then incubating in the dark. The cell-bound dye was then extracted with 200 μL of 10 mM Tris buffer solution (pH 10.5), and its optical density was measured using a multiwall plate reader at 560 nm. The viable cell population was calculated using the following formula: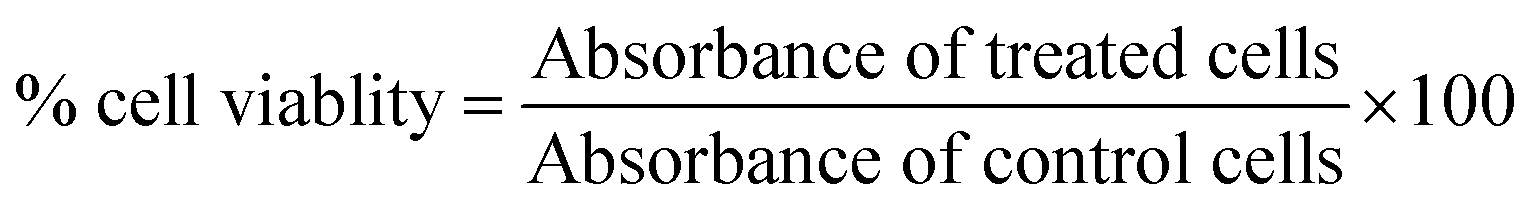
Antibacterial activity of G-Fe3O4
E. coli were procured from K.C. College, Mumbai, India. The cultures were initially isolated from patients infected with water-borne disease. This culture has been sub-cultured for the last 20 years and hence is assumed to have very low virulence. Nutrient broth media was procured from HiMedia. All the chemicals were purchased from Sigma-Aldrich and used without further purification.Results and discussion
Structural and morphological analysis
The phase composition and structures of G–Fe3O4 nanocomposites were examined using X-ray powder diffraction and the corresponding pattern is shown in Fig. S1.† In the pattern of GO (Fig. S1a†), a diffraction peak at 11.08° is observed, which corresponds to the (001) plane of graphene oxide structure. In Fig. S1b,† natural graphite shows diffraction peaks at 26.4°, 44.3° and 54.5° corresponding to the hexagonal lattice of the (002), (101) and (004) planes, respectively. The major peak is at 26.4°, which corresponds to a basal spacing of d002 = 3.38 Å. The typical process is the reduction of GO to graphene during the solvothermal process. After reduction, the peak at 11° completely disappeared and a weak hump (*) could be observed in Fig. S1(c).† In Fig. S1c,† G–Fe3O4 composite with diffraction peaks at 2θ values of 30.6°, 36.1°, 43.7°, 57.6°, 63.1° and 74.7° correspond to (220), (222), (400), (511), (440) and (533) planes of Fe3O4, respectively. This matches with the standard JCPDS no. 65-3107. The results indicate the existence of Fe3O4 in the composite material and clearly show the reduction of GO peak in the composite material. The disappearance of the peak at 2θ = 11° clearly indicated that GO was reduced to graphene. The absence of a sharp peak at 26° indicates that it is not graphite but should be graphene. The peak at 2θ = 26° pertaining to graphene is not as intense because it is in the form of a composite and the Fe3O4 phase dominates the graphene layers. The graphene peak was not predominant because the stacking of graphene sheets in G–Fe3O4 was disordered and exfoliated to a large extent.25,26Morphological characterization of G-Fe3O4
Fig. 1 shows the surface morphology of G–Fe3O4 at different magnifications. The TEM images of G–Fe3O4 reveal that the product consists of a large quantity of Fe3O4 spheres with sizes ranging from 200 to 250 nm. After combination with graphene to form a G–Fe3O4 composite, the Fe3O4 spheres are decorated and firmly anchored on the wrinkled graphene layers with a high density, as shown in Fig. 1. Note that the pleated structure of graphene may favour the hindrance of the Fe3O4 spheres from agglomeration and enable their good distribution on graphene, whereas the Fe3O4 spheres serve as a stabilizer to separate graphene sheets against aggregation. In addition, the Fe3O4 spheres are observed to be porous in nature, which will further help in the adsorption process.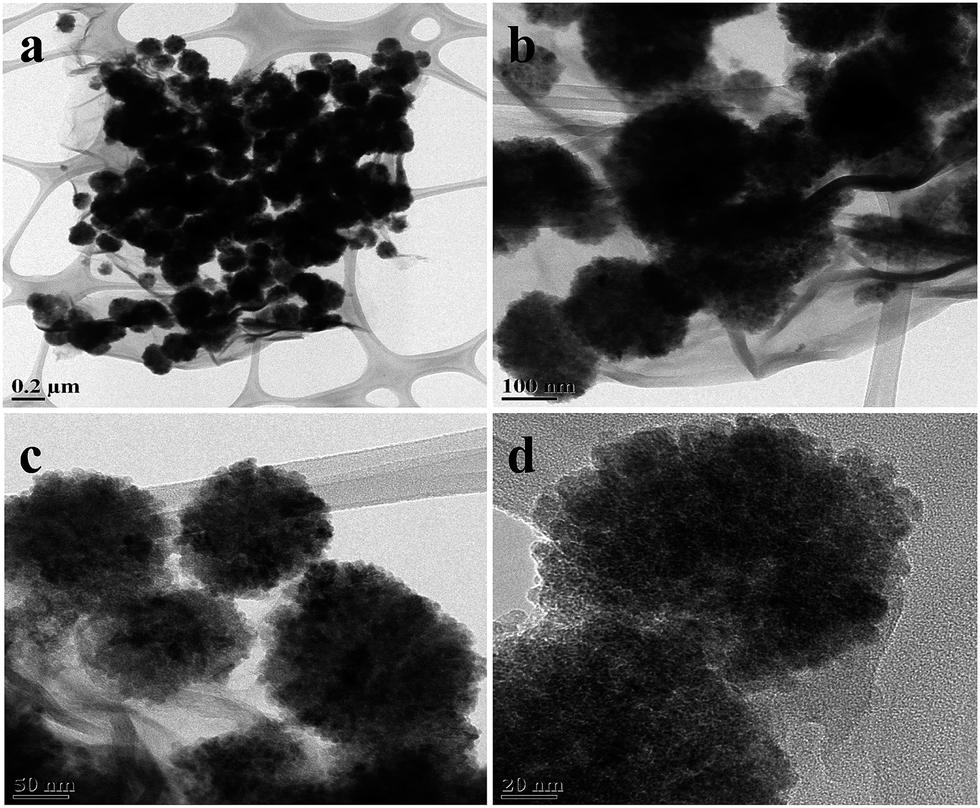 | ||
| Fig. 1 TEM images of G–Fe3O4 nanocomposites. | ||
Further, dynamic light scattering measurements (DLS) were recorded, and the results showed that the nanoparticles are monodispersed (Fig. S2a†). The hydrodynamic sizes of the particles were found to be around 500 nm. In general, the hydrodynamic diameter takes into consideration factors such as solvation and other effects, and hence will be generally higher than that measured by other methods.27 The sizes of the particles were further confirmed by SEM analysis, which showed the dispersion of Fe3O4 particles on graphene sheets (Fig. S2b†).
TGA–DTA/BET analysis
Fig. S3† shows the TGA curve of the as-prepared G–Fe3O4 composite with a minor weight loss from 50 °C to 100 °C (∼10%) and major weight loss from 270 °C to 450 °C (∼40%). After this, no weight loss was obtained up to 1000 °C. A minor weight loss was observed due to adsorbed water and loss of moisture in the as-prepared material. A major weight loss was due to graphene and Fe3O4 composite. Apparently, in DTA curve, two main exothermic peaks were obtained at 290 °C and 450 °C, which relate to the thermal decomposition of ferrites along with carbon.The N2 adsorption–desorption isotherm was used to determine the porous capacity of graphene–Fe3O4 (Fig. S4a†). According to IUPAC (International Union of Pure and Applied Chemistry) classification, the isotherms show typical type IV and type H3 hysteresis loop. This behaviour indicates the predominance of mesopores. The mesoporous nature is also evident from the sharp increment in adsorption volume above the relative pressure of 0.8. The specific surface area of the composite from BET analysis was found to be 27.38 m2 g−1. The Barrett–Joiner–Halenda (BJH) method was used to calculate the pore size on the desorption branch of the N2 adsorption–desorption isotherm. The pore volume versus pore diameter curve (Fig. S4b†) shows a sharp peak at 3.7 nm along with a broad distribution of mesopores. This behaviour also confirms the predominance of mesopores in G–Fe3O4.28
Adsorption parameters
To further explore the adsorption property of the prepared materials, a batch adsorption experiment was conducted; the corresponding adsorption parameters of lead ions onto G–Fe3O4 nanocomposites are shown in Fig. S5.† Fig. S5a† shows the removal efficiency of lead ions onto G–Fe3O4 surfaces, as a function of contact time. It was noted that the adsorption of Pb increased quickly with an increase in contact time, and at a certain period it reached an equilibrium state. It was seen that adsorption is rapid due to the availability of numerous active sites on the adsorbent surface at the initial stage. This fast adsorption may be due to the special one-atom-thick layered structure of GO. In addition, because it contacts Pb ions in the aqueous solution, adsorption occurs immediately due to higher driving force to enable the faster transfer of Pb ions to the active sites on the surface of G–Fe3O4. With further increase in time, the diminishing availability of the remaining active sites and decrease in the driving force makes the adsorption process slow, and thus equilibrium is achieved much later. Thus, the adsorption rate decreases. The equilibrium time increases with increasing initial Pb ion concentration. It can be concluded from Fig. S5a† that it took about 100 min to reach adsorption equilibrium. From 120–180 min, the concentration of the ions remained unchanged.Fig. S5b† shows the amount of adsorbed ions as a function of the initial concentration from aqueous solution. The initial concentration provides an important driving force to overcome the mass transfer of Pb ions between the aqueous and solid phases; hence, a higher initial concentration of Pb ions may increase adsorption capacity. In this experiment, the following concentrations (10, 20, 30, 40, 50 and 60 mg L−1) were chosen at pH 5, equilibrium time of 120 min and T = 310 K. With an increase in ion concentration, the percentage of ion adsorption increased. About 99% of the Pb ions were adsorbed onto G–Fe3O4 surface after 120 min for an initial Pb concentration of 50 mg L−1. Fig. S5c† shows the variation of adsorption capacity of Pb ions by G–Fe3O4 at various pH values. The adsorption capacity increases with an initial pH of 3 and reaches a maximum at pH 7. The effect of pH on the adsorption percentage of Pb ions on the adsorbent G–Fe3O4 were studied at varying pH values over the range of 3–8 using the same concentration of ions; the results are displayed in Fig. S5c.† Although a maximum uptake was noted at pH 8, as the pH increased more than 7, the metal ion started to precipitate. Therefore, no experiment was conducted at pH > 7. The increase in adsorption capacity at pH > 7 could be attributed to both the adsorption of ions onto the surface of adsorbent and precipitation. Therefore, the optimal pH was observed to be 5 for the adsorption of Pb ions. By further increasing the pH adsorption decreased, which was probably due to the formation of Pb hydroxides and chemical precipitation.
Fig. S5d† shows the adsorption% of Pb ions onto G–Fe3O4 matrix as a function of temperature. The percentage-adsorption experiments were conducted at 300, 310 and 320 K to investigate the effect of temperature with an initial concentration of Pb ions of 20 mg L−1, adsorbents dosage of 25 mg L−1 and pH 5. When temperature was increased from 300 to 320 K, the percentage of adsorption of Pb ions increased from 85.2% to 87.2%. An increase in the amount of equilibrium adsorption of Pb ions with an increase in temperature may be explained by the fact that the adsorbent sites were more active at higher temperatures. This condition shows that adsorption occurs more as a physical rather than a chemical process.
Adsorption isotherm study
Developing an appropriate isotherm model for adsorption is essential to design and optimize an adsorption process. Several isotherm models have been developed for evaluating the equilibrium adsorption of compounds from solutions such as Langmuir,29,30 Fruendlich and Dubinin-Radushkevich. The experimental results of this study were fitted with two models. The equilibrium adsorption isotherms are important for determining the adsorption capacity of Pb ions and diagnose the nature of adsorption onto G–Fe3O4 surface.31The equilibrium concentration adsorption capacity of adsorbent was calculated as:
 | (2) |
The adsorption data can then be correlated with Langmuir and Freundlich isotherm model equations. In this study, two classical adsorption models were employed to describe the ion adsorption equilibrium. The Langmuir isotherm is valid for monolayer adsorption onto a surface with a finite number of identical sites.
The Langmuir and Freundlich adsorption isotherm models can be expressed as follows:
 | (3) |
 | (4) |
The slope and intercept of the linear Freundlich equation are 1/n and ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KF, respectively. Fig. 2 shows the adsorption isotherm of Pb ions onto G–Fe3O4 surfaces at 300 K, 310 K and 320 K. From the graph, it can be seen that the adsorption capacity increased with an increase in temperature, indicating an endothermic reaction. We observed that the adsorption percentage increased along with an increase in Ce.
KF, respectively. Fig. 2 shows the adsorption isotherm of Pb ions onto G–Fe3O4 surfaces at 300 K, 310 K and 320 K. From the graph, it can be seen that the adsorption capacity increased with an increase in temperature, indicating an endothermic reaction. We observed that the adsorption percentage increased along with an increase in Ce.
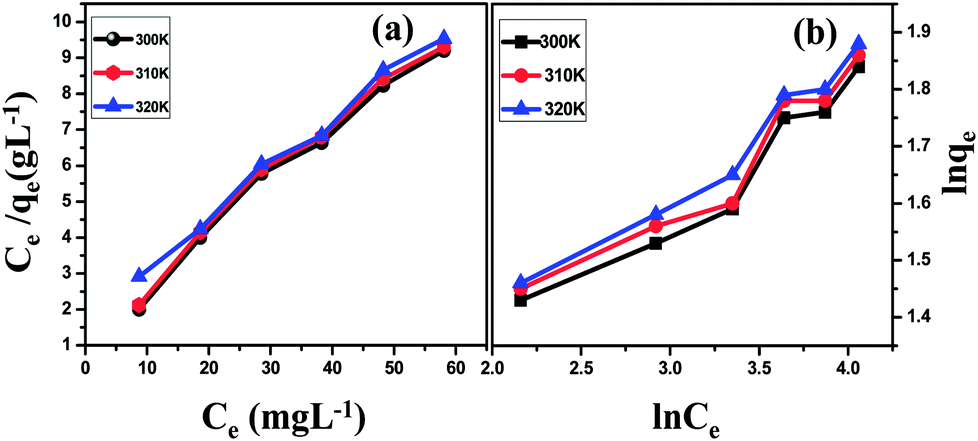 | ||
| Fig. 2 Percentage of adsorption isotherm of Pb ions onto G–Fe3O4 surfaces: (a) Langmuir isotherm; (b) Freundlich isotherm. Initial concentration of the ion was 10, 20, 30, 40, 50, and 60 mg L−1; pH 5; contact time, 120 min and adsorbent dosage 25 mg L−1. | ||
Two models, the Langmuir and Freundlich isotherm models, have been adopted for Pb ion adsorption onto G–Fe3O4 at different temperatures and at different pH values in the experiment. The relative parameters were calculated from the equation and are listed in Table 1. It can be seen from the R2 values that the adsorption isotherms can be simulated well by the two models. The Table suggests that the adsorption of Pb on G–Fe3O4 is mainly a monolayer type.
| Temp (K) | Langmuir | Freundlich | ||||
|---|---|---|---|---|---|---|
| Kd | qm (mg g−1) | R2 | KF | n | R2 | |
| 300 | 0.12 | 69 | 0.998 | 4.2 | 0.23 | 0.9328 |
| 310 | 0.1 | 69 | 0.992 | 26.96 | 0.24 | 0.9248 |
| 320 | 0.1 | 67 | 0.9818 | 10.86 | 0.27 | 0.9686 |
It can be inferred from the R2 values given in Table 1 that the Langmuir isotherm fits the experimental data better than the Freundlich isotherm. It was also noted that adsorption is a monolayer process and that the adsorption of all species requires equal activation energy. From the table it is clear that at different temperatures there is change in qm and Kd values; the maximum adsorption capacity qm obtained was 69 mg g−1.
Thermodynamic parameters
Thermodynamic parameters provide additional in-depth information regarding the inherent energetic changes involved during adsorption. To assess thermodynamic parameters, the adsorption isotherm of Pb ions onto G–Fe3O4 surfaces were measured at 300, 310 and 320 K, and the changes in thermodynamic parameters of standard Gibbs free energy of adsorption (ΔG°), standard enthalpy (ΔH°) and standard entropy (ΔS°) were calculated from the variation of thermodynamic equilibrium constant, Ko, with a change in temperature.The standard Gibbs free energy of adsorption, ΔG°, is
|
ΔG° = −RTlnKo
| (5) |
|
RTlnKo = TΔS° − ΔH°
| (6) |
From Table 2, it is clear that the value of change in standard enthalpy is positive and Gibbs free energy is negative, showing that the adsorption of Pb ions onto G–Fe3O4 adsorbents is endothermic. Consequently, it can be seen that there is a change in the adsorption as temperature increases, and here it is seen that the adsorption is same at 300 and 310 K.
| Temp (K) | ΔG° | ΔH° (kJ mol−1) | ΔS° (kJ mol−1) |
|---|---|---|---|
| 300 | −1643 | ||
| 310 | −4796 | 255.8 | 15.908 |
| 320 | −9686 |
A positive standard enthalpy change suggests that the interaction of Pb ions with G–Fe3O4 is endothermic, which is supported by the increasing adsorption of Pb with increase in temperature. A comparison has been made of the adsorption capacity for various adsorbents cited in the literature for the removal of Pb ions (Table 3). It is clear from the table that our G–Fe3O4-based adsorbent has the maximum Pb ion adsorption capability.
Photo-Fenton degradation of dye
Further work was carried out for probing the role of this material towards dye degradation applications. Dye adsorption on the catalyst surface is a prerequisite for efficient dye removal. The adsorption of methylene blue dye was studied within a 30 min time interval using UV-vis spectroscopy. Fig. S6† shows that the adsorption of dye is quite rapid in the first 30 min, and then gradually rises with increase in adsorption time. After 180 min, the amount of dye removed from the suspension is calculated using the following equation:
 | (7) |
Graphene shows much better removal efficiency compared with the composite of the same amount (0.2 g L−1), which can be attributed to the large surface area of graphene sheets. In addition, π–π stacking between the dyes and π-conjugated regions of the sheets can offer more active adsorption sites and catalytic reaction centers.36 The photocatalytic property of the composite is measured by the degradation of the dye in the presence of visible light (Fig. 3). Before light irradiation, the suspension of the catalyst and the dye was stirred for 180 min in the dark to reach the adsorption–desorption equilibrium between the catalyst and the dye. It has been observed from Fig. 3(a and c) that upon the addition of H2O2 in the presence of visible light, the degradation rate increases rapidly. For comparison, the effect of H2O2 in the dark on G–Fe3O4 composite is also shown in a graph (Fig. 3(b)); an enlarged view of dye degradation is also shown as an inset in Fig. 3. The enhancement in dye degradation can be attributed to the synergistic interaction between GO and Fe3O4 nanoparticles. The electron transfer between NPs and GO sheet facilitates the reduction of Fe3+ to Fe2+ ion. Subsequently, Fe2+ would react with H2O2 to produce hydroxyl radicals (OH˙). The hydroxyl radicals produced as a result of Fenton/photo-Fenton processes attacks the dye adsorbed on the surface of the catalyst and degrade it to CO2 and H2O.37,38
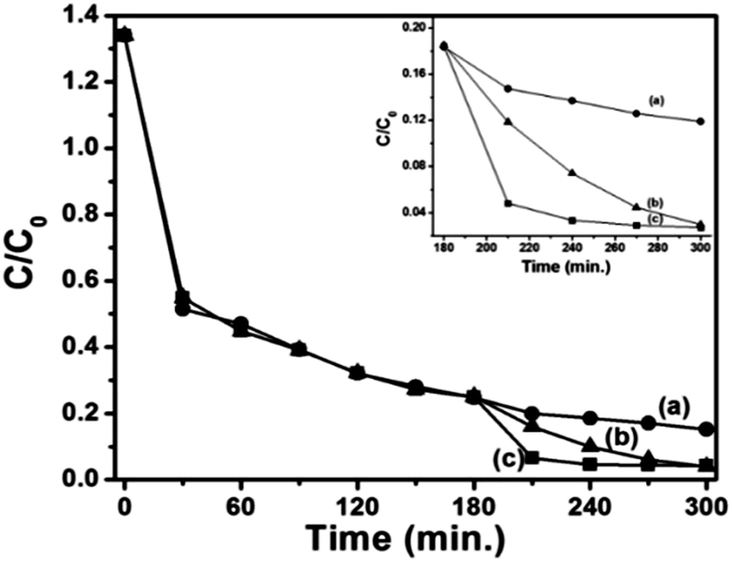 | ||
| Fig. 3 Photo-Fenton degradation spectra of G–Fe3O4 composite at a concentration of 0.2 g L−1 in the presence of (a) visible light, (b) H2O2 and (c) visible light and H2O2. (Inset shows the expanded view). | ||
For the recycling of G–Fe3O4 composite, the absorbent was separated, washed and then reused. The recycled adsorbed behaviour is shown in Fig. S7.† It was observed that the removal efficiency of the dye was over 95% up to 5 cycles. The photocatalytic activity of the recycled catalyst was also investigated, and the results are shown in Fig. S7.† There was no noticeable change in the photocatalytic activity of the recycled catalyst after five cycles under visible light irradiation, indicating that the magnetically separable photocatalyst is stable and effective for the degradation of organic pollutants in water.
Antibacterial activity of G -Fe3O4
Fig. 4 shows that about 22.66% of the cells were only viable when treated with G–Fe3O4, whereas with graphene about 65.33% of the cells were viable. The two activities were further examined, i.e., concentration and time-dependent behaviour of G–Fe3O4 on E. coli cells. Different concentrations of G–Fe3O4 (0, 25, 50, 100, 200 ppm) dispersions were incubated with E. coli cells for 60 min and then plated for investigating cell viability. Only E. coli cells and graphene were controls. | ||
| Fig. 4 Cell viability of E. coli when treated with graphene and G–Fe3O4. | ||
It was observed that 25 ppm G–Fe3O4 could inhibit 43% of the cells because the recorded cell viability according to Fig. S8(a)† is 57%. In addition, 100 ppm was sufficient to inhibit bacteria for cell viability at 26.84% E. coli cells.
On the other hand, in a time-dependent study, we observed that when 100 ppm of G–Fe3O4 dispersion was treated with E. coli cells for various time intervals from 0 to 90 min, 60 min was sufficient to kill bacteria, showing 23.74% cell viability (Fig. S8(b)†). The loss of E. coli viability progressively decreased with an increase in G–Fe3O4 concentration. E. coli viability almost decreased from 57% by 25 ppm G–Fe3O4 to 21% with 200 ppm.
The majority of E. coli cells were killed after incubation with G–Fe3O4 at a concentration of 100 ppm. In a similar manner, cell viability w.r.t. time from 0–90 min drastically reduced from 64.66% to 19.66%.
Destruction of bacterial membrane
FE-SEM was used to illustrate the interaction between G–Fe3O4 and E. coli. In Fig. 5(a), E. coli cells as a control show the entire cell wall without any rupture, whereas Fig. 5(b) shows complete disintegration of the cell wall. Spikes created on the E. coli cells are a clear indication that G–Fe3O4 interacted with the cell wall. This kind of cell wall rupture is irreversible damage induced by direct contact with G–Fe3O4. | ||
| Fig. 5 (a) FE-SEM images of E. coli cells control and (b) rupture by G–Fe3O4. | ||
Fig. S9† shows the biocompatibility study. It was essential to understand whether G–Fe3O4, if leached during water purification, would cause harm to normal cells. As shown, graphene has less cell compatibility of about 54.11% in 2 mg mL−1, whereas at the same concentration compatibility for G–Fe3O4 is about 92.99%. Presence of Fe3O4, which supposedly is biocompatible, has been a good support with graphene as a biocompatible material.
Biochemical assay of total cellular protein degradation due to G -Fe3O4
Degradation of protein due to the effect of graphene and G–Fe3O4 was performed by Folin-Lowry test. Table 4 (in ESI†) also indicates a reduction in protein content when E. coli was treated with graphene and G–Fe3O4 individually. The reduction in total protein content was found to be approximately 86.66% in the case of G–Fe3O4. The total cellular protein degraded drastically to 86.66 when treated with G–Fe3O4, as observed from Table 2, whereas the control showed about 236% of protein. Perhaps this is why only 22.66% of the E. coli could survive (i.e., 78% mortality) when exposed to 100 ppm G–Fe3O4 nanocomposite, suggesting that microbial death is due to the degradation of total cellular protein.Total protein assay (by SDS-PAGE methods) of E. coli due to G-Fe3O4
Fig. 6 depicts the degradation of protein as analyzed by SDS-PAGE electrophoresis upon treating E. coli cells with G–Fe3O4. It is clear from the protein degradation results that stress-mediated protein degradation of cell membrane left only one higher density protein intact, whereas the rest of the proteins were denatured, unlike proteins treated with graphene alone or control. | ||
| Fig. 6 Protein degradation by G–Fe3O4. | ||
Glutathione oxidation due to G -Fe3O4 treatment
Disregulation of H2O2 pathway in a cellular membrane causes oxidative stress. To examine oxidative stress, in vitro GSH oxidation was mediated.GSH is a tripeptide with thiol groups, and is an antioxidant in bacteria at a concentration ranging between 0.1 and 10 mM. GSH can prevent damages to cellular components caused by oxidative stress. Thiol groups (–SH) in GSH can be oxidized to disulfide bonds (–S–S–), which converts GSH to glutathione disulfide. GSH has been used as an oxidative stress indicator in cells. The Ellman's assay is able to quantify the concentration of thiol groups in GSH. When 0.4 mM GSH was incubated with 100 ppm G–Fe3O4, the oxidation of GSH gradually advanced, extending the reaction time by up to 60 min. Fig. 7 shows that the fraction of GSH oxidized by G–Fe3O4 increases in comparison with graphene. Comparably, G–Fe3O4 has significantly higher oxidation reactivity up to 89.32% more than graphene at the same reaction time and concentration of 60 min in 125 μg mL−1 of G–Fe3O4. The oxidation of GSH indirectly confirms that G–Fe3O4 is capable of mediating ROS-independent oxidative stress toward bacterial cells.
 | ||
| Fig. 7 Oxidative stress release by E. coli due to G–Fe3O4. | ||
Thus, the prepared composite is effective for multipurpose applications due to the combined advantages of Fe3O4 and graphene.
Conclusions
G–Fe3O4 was synthesized by a simple one-step solvothermal route from graphene oxide and iron oxide nanoparticles. This nanostructure serves as an efficient adsorbent for the removal of heavy metal ions from waste water. The maximum adsorption capacity of G–Fe3O4 was 17 mg g−1 at an initial Pb concentration of 20 mg L−1 and temperature of 310 K, indicating that G–Fe3O4 is effective for the adsorption of Pb(II) ions. The experimental data fitted well with the Langmuir isotherm model. The monolayer adsorption capacity of Pb by G–Fe3O4 was found to be 69 mg g−1 at 310 K at pH 5. Thermodynamic investigations indicated that the adsorption reaction was spontaneous and endothermic. Results indicate that such materials could be used as adsorbents for the removal of heavy metal ions. Photodegradation of methylene blue dye was successfully achieved up to 5 cycles. Antibacterial activity depicts that only 22.33% of the cells were viable when treated with G–Fe3O4. Further protein degradation confirmed that a higher molecular weight protein was not denatured, whereas the rest of the proteins as compared with E. coli had disintegrated. This was due to the release of some ROS and disintegration of disulphide bonds.Acknowledgements
Dr Pratap Kollu acknowledges DST-INSPIRE Faculty grant and authors acknowledge SAIF/CRNTS and Central surface analytical facility of IIT Bombay. Also the authors gratefully acknowledge VIT University, Vellore for supporting this work under the research associate fellowship.References
- A. K. De, Environmental Chemistry, Wiley Eastern Ltd., New Delhi, 2nd edition, 1992 Search PubMed
.
- S. Singh, K. C. Barick and D. Bahadur, J. Hazard. Mater., 2011, 193(3), 1539–1547 CrossRef PubMed
- N. A. Babarinde, J. O. Babalola and R. A. Sanni, Int. J. Phy. Sci., 2006, 1, 23–26 Search PubMed
- A. Saeed, M. Iqbal and M. W. Akhtar, J. Hazard. Mater. B, 2005, 117, 65–73 CrossRef CAS PubMed
- T. A. Davis, B. Volesky and A. Mucci, Water Res., 2003, 37, 4311–4330 CrossRef CAS
- G. McKay and Y. S. Ho, Water Res., 1999, 33, 578–584 CrossRef
- B. H. Hameed, A. A. Ahmad and N. Aziz, Desalination, 2009, 247, 551–560 CrossRef CAS PubMed
- V. K. Gupta, A. Mittal, R. Jain, M. Mathur and S. Sikarwar, J. Colloid Interface Sci., 2006, 303, 80–86 CrossRef CAS PubMed
- S. Liu, T. Helen Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5(9), 6971–6980 CrossRef CAS PubMed
- S. Singh, K. C. Barick and D. Bahadur, Nanomater. Nanotechnol., 2013, 3, 1–19 CrossRef PubMed
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed
- M. Ghaedi, A. Hassanzadeh and S. N. Kokhdan, J. Chem. Eng. Data, 2011, 56, 2511–2520 CrossRef CAS
- T. S. Sreeprasad, S. M. Maliyekkal, K. P. Lisha and T. Pradeep, J. Hazard. Mater., 2011, 186, 921–931 CrossRef CAS PubMed
- J. Xu, L. Wang and Y. Zhu, Langmuir, 2012, 28, 8418–8425 CrossRef CAS PubMed
- S. Gupta, T. S. Sreeprasad, S. M. Maliyekkal, S. K. Das and T. Pradeep, ACS Appl. Mater. Interfaces, 2012, 4, 4156–4163 CAS
- S. Wang, H. Sun, H. M. Ang and M. O. Tade, Chem. –Eng. J., 2013, 226, 336–347 CrossRef CAS PubMed
- M. Machida, T. Mochimaru and H. Tatsumoto, Carbon, 2006, 44, 2681–2688 CrossRef CAS PubMed
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 55406 CrossRef
- Z. Wu, D. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS
- J. Hu, L. Zhong, W. Song and L. Wan, Adv. Mater., 2008, 20, 2977–2982 CrossRef CAS
- Y. M. Zhai, J. F. Zhai, M. Zhou and S. J. Dong, J. Mater. Chem., 2009, 19, 7030–7035 RSC
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5, 6971–6980 CrossRef CAS PubMed
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS
- J. P. Ruparelia, S. P. Duttagupta, A. K. Chatterjee and S. Mukherji, Desalination, 2008, 232, 145–156 CrossRef CAS PubMed
- B. Li, Y. Fu, H. Xia and X. Wang, Mat. Lett., 2014, 122, 193–196 CrossRef CAS PubMed
- Z. S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed
- J. K. Lim, S. P. Yeap, H. X. Che and S. C. Low, Nanoscale Res. Lett., 2013, 8, 381 CrossRef PubMed
- K. C. Barick, S. Singh, M. Aslam and D. Bahadur, Microporous Mesoporous Mater., 2010, 134, 195–202 CrossRef CAS PubMed
- H. Y. Koo, H. J. Lee, H. A. Go, Y. B. Lee, T. S. Bae, J. K. Kim and W. S. Choi, Chem.–Eur. J, 2011, 17, 1214–1219 CrossRef CAS PubMed
- L. Ren, S. Huang, W. Fan and T. Liu, Appl. Surf. Sci., 2011, 258, 1132–1138 CrossRef CAS PubMed
- T. Liu, Y. Li, Q. Du, J. Sun, Y. Jiao, G. Yang, Z. Wang, Y. Xia, W. Zhang, K. Wang, H. Zhu and D. Wu, Colloids Surf., B, 2012, 90, 197–203 CrossRef CAS PubMed
- Y. H. Li, Z. Di, J. Ding, D. Wu, Z. Luan and Y. Zhu, Water Res., 2005, 39, 605–609 CrossRef CAS PubMed
- Z. Reddad, C. Gerente, Y. Andres and P. Cloirec Le, Environ. Sci. Technol., 2002, 36, 2067–2073 CrossRef CAS
- B. Salih, A. Denizli, C. Kavakli, R. Say and E. Piskin, Talanta, 1998, 77, 1147–1154 Search PubMed
- C. H. Lai and C. Y. Chen, Chemosphere, 2001, 44, 1177–1184 CrossRef CAS
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380 CrossRef CAS PubMed
- S. Q. Liu, B. Xiao, L. R. Feng, S. S. Zhou, Z. G. Chen, C. B. Liu, F. Chen, Z. Y. Wu, N. Xu, W. C. Oh and Z. D. Meng, Carbon, 2013, 64, 197–206 CrossRef CAS PubMed
- S. Guo, G. Zhang, Y. Guo and J. C. Yu, Carbon, 2013, 60, 437–444 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02913e |
| ‡ Current address: Thin Film Magnetism group, Cavendish Laboratory, Department of Physics, University of Cambridge, Cambridge CB3 0HE, UK. Email: pk419@cam.ac.uk |
| This journal is © The Royal Society of Chemistry 2014 |