Novel nickel–palladium catalysts encased in a platinum nanocage†
Balachandran Jeyadevan*a,
Jhon L. Cuyaa,
Yoshinori Inouea,
Kozo Shinodab,
Takashi Itoc,
Derrick Mottd,
Koichi Higashimined,
Shinya Maenosonod,
Takatoshi Matsumotob and
Hiroshi Miyamuraa
aDepartment of Material Science, University of Shiga Prefecture, 2500 Hassaka Cho, Hikone 522-8533, Shiga, Japan. E-mail: jeyadevan.b@mat.usp.ac.jp
bInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Sendai, Japan
cFrontier Research Institute for Interdisciplinary Sciences, Tohoku University, Sendai, Japan
dSchool of Material Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa Prefecture 923-1292, Japan
First published on 2nd June 2014
Abstract
Novel Ni–Pd nanocubes of a few tens of nm in size encased in a Pt cage have for the first time been synthesized using a long-chain alcohol reduction process. The unique distribution of Pt atoms in these particles holds the key for the design of high turnover catalysts for the future.
1. Introduction
In recent years strategic initiatives encouraging the development of alternate functional materials to replace those based on rare earths and precious metals have been introduced in developed countries to overcome the limited and geographically constrained supply of these materials. Precious transition metals such as Pt and Pd have demonstrated outstanding potential as catalysts in chemical, electrocatalytic, automotive and other key industries, both as the pure metal and in the form of alloys.1,2Although considerable progress has been made in the preparation of precious transition metal-based colloidal nanostructures, research to replace these metals with abundantly available alternative elements has not met with any great success. This has forced researchers to look to alloying as a method for optimizing their use and developing techniques to preserve their availability through recycling. It is well known that alloying with a second metal can significantly improve the catalytic properties of the original metal, and the synthesis of multi-metallic nanoparticles (NPs) has become a promising topic.3–5 Alloy NPs based on Pt–Pd, Pd–Ni and Pt–Ni have attracted considerable attention,6–8 and improved catalytic activity compared with Pt alone has been reported in the case of core–shell structured Pt–Pd NPs.9 The search for techniques that might enable the formation of new structures of high catalytic activity using smaller amounts of Pt is particularly being pursued. In addition, attempts have been made to improve the efficiency and selectivity of Pt-based NPs for specific reactions by the engineered exposure of specific crystal facets.10–13
In all cases, these studies emphasize that fine control over compositions and surface structures is necessary for the creation of high-throughput and cost-effective Pt-based nanostructures. Following this line of thought, the present authors have attempted the synthesis of magnetic Pt-based NP alloys such as Fe–Pt,14 Co–Pt15 and Ni–Pt,16 which reduce the net amount of platinum required and also impart magnetic properties, and which enables the collection of these materials after use and assists in the recovery of the precious metals. In the case of Fe–Pt NPs synthesized by the polyol process, deposited on activated carbon and then heat-treated to convert the product to the face-centred tetragonal structure, these have been demonstrated to offer a possible alternative catalytic electrode material for polymeric electrolyte cells.17 On the other hand the synthesis of Ni NPs,18 as well as novel cubic-shaped Ni–Pt NPs,16 has also been achieved through an alcohol reduction technique. Furthermore, in a preliminary evaluation of the catalytic performance of cubic-shaped Ni–Pt NPs in the hydrogenation of 1-octene to octane, Ni91Pt9 NPs (10 nm cubes) have performed in a comparable manner to Pt (3.5 nm) metal NPs, once the specific surface area is taken into consideration.
The similarity of properties such as atomic radius, crystal structure and electronegativity between Pt and Pd, and the use of these metals as catalysts in the automobile industry for the enhancement of the activity of carbon dioxide and hydrocarbons, has motivated the synthesis of Ni–Pd–Pt NPs by extending the already established process for the preparation of Ni–Pt NPs to the synthesis of cubic-shaped NPs consisting of Pt, Pd and Ni metals. In the present study we report the synthesis of novel Pd-core–Pd-Ni shells holding Pt nanocage particles; and these look very promising for application as catalysts.
2. Experimental
2.1 Materials
Nickel(II) acetate tetrahydrate [Ni(CH3COO)2·4H2O], 98% purity, dihydrogen hexachloroplatinate hexahydrate (H2PtCl6·6H2O), 98.5%, and palladium acetylacetonate [Pd(C5H7O2)2], 99%, were purchased from Sigma-Aldrich and formed the metal sources. Solvents such as 1-heptanol (98%), methanol (99.8%), 2-propanol (99.9%), 1-octene (99.9%), octane (98.0%), toluene (99.5%) and oleylamine [CH3(CH2)7CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)7CH2NH2], 70%, were purchased from Wako Pure Chemicals Ltd., Japan. Commercial reagents were used without further purification.
CH(CH2)7CH2NH2], 70%, were purchased from Wako Pure Chemicals Ltd., Japan. Commercial reagents were used without further purification.
2.2 Synthesis of Ni–Pt–Pd nanoparticles
In a typical procedure to synthesize Ni–Pd–Pt NPs, 3.8 mM of the nickel salt was totally dissolved in 5 mL methanol using ultrasonication and then mixed with 100 mL 1-heptanol containing 42 mM oleylamine, 0.1 mM Pd(C5H7O2)2 and 0.85 mM H2PtCl6·6H2O, before heating at 170 °C for 40 min. The resulting NPs were collected using a magnet and washed with a mixture of methanol and toluene to remove unreacted compounds and excess oleylamine. The NPs were finally redispersed in toluene.2.3 Characterization
The powder X-ray diffraction (XRD) patterns of the samples were determined using an X-ray diffractometer (Philips-Xpert) with Cu-Kα radiation to characterize the crystal phases present in the powder. The size and morphology of the particles were assessed using a transmission electron microscope (TEM; Hitachi H8100) at 200 kV. The samples for TEM measurement were prepared by depositing a toluene dispersion of Ni–Pt–Pd particles on amorphous carbon-coated grids. In addition, high-angle annular dark field (HAADF) microscopy, coupled with scanning transmission electron microscopy (STEM) and energy-dispersive spectroscopy (EDS) elemental mapping, were performed on a JEOL JEM-ARM200F instrument operated at 200 kV with a spherical aberration corrector; the nominal resolution was 0.8 Å. STEM-HAADF imaging and EDS mapping analysis allowed us to visualize clearly the relative positions of Ni, Pd and Pt within the individual Ni–Pd–Pt alloy NPs.In order to investigate the local atomic environmental structure around certain elements, the extended X-ray absorption fine structure (EXAFS) spectrum of the sample for the Pt L3 absorption edges was recorded using an in-house X-ray absorption spectrometer, the Rigaku R-XAS Looper (Tokyo, Japan). In the spectrometer, the demountable X-ray tube with Mo target as the white X-ray source and the Si(400) Johansson-type bent single crystal as the monochromator crystal were employed. The experiments at the Pt L3 absorption edges were carried out in the transmission mode using samples diluted with boron nitride powder or pelletized, and in the fluorescence yield mode using the samples pressed without dilution. The data processing of the measured X-ray absorbance spectra was conducted using the program REX2000 v. 2.5.9 produced by Rigaku.
The local chemical environment of the constituent atoms was carried out on a Shimadzu Kratos AXIS-ULTRA delay-line detector high-performance X-ray photoelectron spectroscopy (XPS) system. The XPS measurement details were as follows. Photoelectrons were excited using monochromated Al Kα radiation. Detection was performed with a delay-line detector and a concentric hemispherical analyzer. The X-ray tube was operated at 150 W. The pass energy of the concentric hemispherical analyzer was 20 eV for narrow-scan spectra. The analyzed area on the specimen surface was 300–700 mm2 and was located in the centre of the irradiated region. For sample preparation, the precipitated NPs were deposited on carbon tape and dried in air. The instrument was operated at a vacuum of 10−8 Torr.
The catalytic properties of Ni–Pd–Pt NPs were examined using the hydrogenation reaction of 1-octene to octane. In this regard, a specific quantity of Pt, Ni–Pd–Pt or acid-treated Ni–Pd–Pt NPs, was introduced into a three-necked glass round flask together with 50 mL 2-propanol and 5 mL 1-octene. The solution was heated at 83 °C under a constant flow of pure hydrogen (99.999%) with mechanical stirring (300 rpm). After refluxing 2 h the solution was cooled and then analyzed by gas chromatography to determine the degree of octane conversion.
3. Results and discussion
To monitor the formation of NPs, sampling was conducted at different points once the reactants had reached the specified temperature. Fig. 1 shows TEM micrographs of samples taken at 20, 30 and 40 min at a reaction temperature of 170 °C. | ||
| Fig. 1 TEM micrographs of Ni–Pd–Pt NPs sampled at reaction times (a) 20, (b) 30 and (c) 40 min. | ||
Although the NPs were less uniformly shaped after 20 min, they became entirely cubic after 30 min reaction. However, once the reaction had progressed further the NPs became irregular in shape and non-uniform in size. At this point we speculated that the precipitation of different crystal phases had occurred over time. We therefore decided to analyze these NPs structurally using X-ray diffraction (Rigaku RINT2000), using Cu-Kα radiation as the incident X-ray source. The results of the measurements are shown in Fig. 2.
 | ||
| Fig. 2 XRD patterns of Ni–Pt–Pd NPs samples withdrawn after (a) 20, (b) 30 and (c) 40 min reaction time at 170 °C. | ||
The XRD patterns showed a sequential shift to the right in the face-centred cubic structure peaks, corresponding to the Pt–Pd(111) peak. The main peak of Pt/Pd is shifted to a higher angle as the reaction progresses. This is due to the increase in the Ni composition of the Ni–Pd–Pt alloy NPs, which will decrease the lattice constant of Pt/Pd. The relationship between the lattice constants derived from the XRD (Ni0.59(PdPt)0.41) and the composition of the NPs evaluated from the EDS analysis (Ni0.72(PdPt)0.28) showed a positive deviation from the ideal Vegard's law. On the other hand, in the case of the Ni(111) peak a slight shift to the left was observed for the sample taken after 20 min reaction time, but within 30 min the diffraction peaks corresponding to both Ni and Pd/Pt had merged into a single peak, suggesting the formation of Ni–Pd–Pt alloys. The lattice constant of the alloy was calculated from the XRD pattern to be approximately 0.367 nm. In this alloy phase the nearest neighbouring interatomic distance is 0.260 nm. After 40 min reaction, in addition to the peak corresponding to the Ni–Pd–Pt alloy, the reappearance of the Ni(111) peak was observed. This was due to the reduction of the Ni ions remaining in the system and their subsequent precipitation on the surface of the cubic particles. Consequently, the particles then lost their shape and became very irregular.
From the above results we were able to postulate that during the initial stages of the reaction Ni, Pd and Pt became alloyed, and that after 30 min reaction the Ni atoms remaining in solution began to precipitate preferentially on the surface of the alloyed NPs. This proposed reaction scheme also explains the reason for the shape and size of the NPs observed after 40 min reaction, shown in Fig. 1(c). Most of the particles synthesized under optimum conditions were nearly cubic (see the inset in Fig. 1(b)). However, when the lengths of the particles are not exactly the same they then look different, and the projected figure becomes complicated since the particles are standing on their corners. The high resolution TEM does not confirm the formation of twining or intergrowth. Although the morphological and structural analyses reveal the formation and growth mechanisms, the local atomic environment within the NPs has yet to be resolved. Thus, in order to analyze the local atomic environmental structure around the Pt atoms, the XAFS spectrum of the sample for the Pt L3 absorption edge was recorded using the in-house X-ray absorption spectrometer, housing a demountable X-ray tube with Mo target as the white X-ray source and a Si(400) Johansson-type bent single crystal as the monochromator. The experiments were carried out in the fluorescence yield mode using the pressed samples.
The FEFF 8.20 code was used for the theoretical calculation of the back-scattering amplitudes and phase shift parameters of scattering photoelectrons and the REX2000 v .2.5.9 program (Rigaku) was used for the analysis of the measured EXAFS spectrum. The correlation peak for the nearest neighbouring pair corresponded to Pt–Ni.16 In the Ni–Pd–Pt alloy, the nearest neighbour corresponded to Pt–Ni, as in the Ni–Pt alloy. The distance of 0.258 nm obtained corresponded well with the value calculated from the XRD pattern. The measured Fourier transforms of k3-weighted EXAFS spectrum for the Ni–Pd–Pt alloy sample is shown in Fig. 3, along with that for the Ni–Pt alloy.16 In the Pt L3 Fourier transform for the Ni–Pd–Pt sample a small atomic pair correlation is indicated at a further distance, as the shoulder of the nearest neighbouring Pt–Ni pair correlation peak. This correlation is not considered as the second nearest neighbouring peak in the FCC alloy, since the observed distance is too far below 0.367 nm, even after taking into account the shift of peak position in the Fourier transform caused by the phase shift of oscillation in the EXAFS spectrum. Thus from the correlation shoulder position, this is considered to be the consequence of Pt–Pt (0.277 nm in pure platinum) or Pt–Pd (0.275 nm in Pt–Pd alloy) in the Ni-diluted alloy. Considering the results obtained from elemental distribution mapping shown later in Fig. 5, the closer correlation peak and the further correlation shoulder in the Fourier transform are in correspondence with the nearest neighbouring Pt–Ni pair in the Ni–Pt alloy located at the shell area of the particle and the Pt–Pt pair in the Pt-rich alloy located at the edge of particle, respectively.
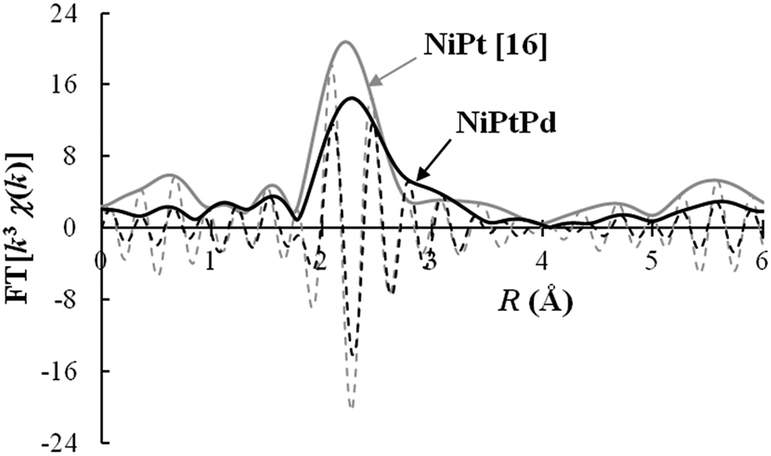 | ||
| Fig. 3 Fourier transforms of k3 weighted EXAFS spectra measured at the Pt L3 absorption edge for the samples of Ni–Pd–Pt (in the present study the reaction time was 30 min) and Ni–Pt.16 | ||
To obtain additional information about the local chemical environment of the constituent atoms, we conducted X-ray photoelectron spectroscopy (XPS) analysis. This was carried out on a Shimadzu Kratos AXIS-ULTRA delay-line detector high-performance XPS system. Fig. 4 shows the high-resolution Ni2p XPS core-level spectrum of the Ni–Pd–Pt alloy NPs. The background subtraction was performed using the Shirley method. The shift in the binding energy (BE) scale due to charging was corrected by internal referencing of the C1s peak (284.4 eV). The Ni2p core levels are split into 2p1/2 and 2p3/2 spin–orbit pairs. To examine the Ni2p XPS spectrum in more depth the Ni2p spectrum was deconvoluted into three different Ni species, including Ni–Pd metal/alloy, oxide/hydroxide, and a structure with an intense satellite signal at high BE adjacent to the main peaks, which could be attributed to multi-electron excitation. After these shake-up peaks were taken into account, the Ni2p3/2 XPS peaks at binding energies of 852.0, 853.8 and 859.7 eV were assigned to Ni–Pd alloy,19 Ni oxide/hydroxide and satellite, respectively. Note that the BEs of Pd3d and Pt4f showed that Pd and Pt were present in a zero-valency metallic state in the Ni–Pd–Pt alloy NPs (ESI, Fig. S1†).
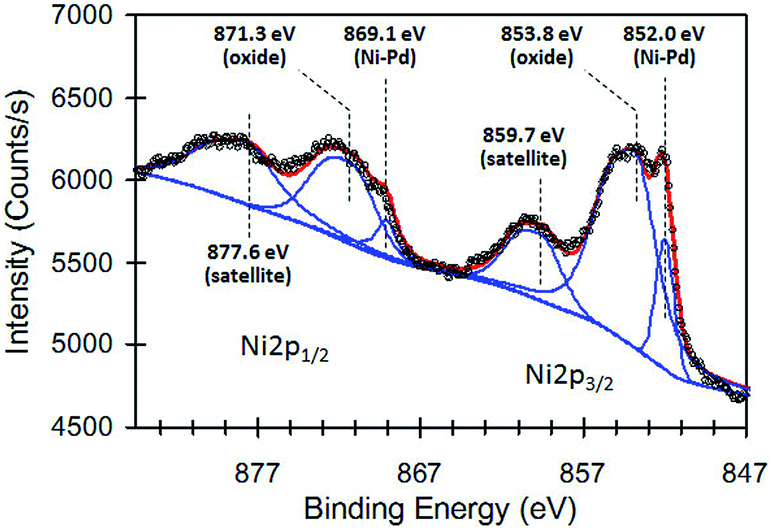 | ||
| Fig. 4 High-resolution Ni 2p XPS core-level spectrum of the Ni–Pd–Pt sample synthesized at 170 °C for 30 min. | ||
Although a considerable amount of information was obtained by the above techniques, a distinction between the behaviour of Pt and Pd was not possible, nor was information available about their role in the formation of the particles or their distribution within them. We therefore attempted an observation of the atomic distribution using STEM. Using HAADF microscopy coupled with STEM and energy-dispersive spectroscopy (EDS), elemental mapping was performed on a JEOL JEM-ARM200F instrument operating at 200 kV with a spherical aberration corrector; the nominal resolution was 0.8 Å. STEM-HAADF imaging and EDS mapping analyses allowed us to visualize clearly the relative positions of Ni, Pd and Pt within the individual Ni–Pd–Pt alloy NPs (Fig. 5).
 | ||
| Fig. 5 (a) STEM-HAADF image, and (b–d) EDS elemental mapping images of the Ni–Pd–Pt alloy NPs; (b) Ni K edge, (c) Pd L edge, and (d) Pt M edge. | ||
Since the heavier Pt atoms (atomic number, 78) give rise to a brighter image than the lighter Ni (28) and Pd (46) atoms in the dark field image, the Pt cage appeared brighter than the Ni–Pd core. When comparing this image with the EDS elemental map, it is seen that the majority of Pt atoms exist at the vertices and sides of the cubic NPs. In addition, it is also seen that the cubic Ni–Pd core consists of a Pd-rich centre and a Ni–Pd alloy outer layer. The spatial distribution of Pd within the NPs suggests that Pd ions are preferentially reduced compared to Pt, and this facilitates heterogeneous nucleation and subsequent co-reduction of Ni and Pd, leading to the formation of a uniform Ni–Pd alloy shell around a Pd core. Surprisingly, Pt ions are not reduced in preference to Pd, nor even Ni. Apart from a recent report on the synthesis of Pt–Pd alloy NPs,6 the one-pot reduction of Pt and Pd is often claimed to form core–shell NPs. In contrast, Pd alloys preferentially with Ni rather than Pt, and this accounts for the vast amount of literature on Pd–Ni alloy formation.7,20 Pt ions are thus not allowed to be incorporated in the Ni–Pd alloy and are finally reduced at the active sites of the Ni–Pd cubes, such as the corners and edges. However, the reason for the absence of separate Pt NPs is not at present clear. Furthermore, the driving force for the formation of the cubic shape of the NPs in the Ni–Pd–Pt ternary systems also needs to be investigated.
Since the Ni–Pt NPs also formed a cubic configuration, we carried out STEM analysis to study the distribution of Pt in these NPs. Here again we were able to identify the presence of Pt at the edges and corners of the cubes (ESI, Fig. S2†). In contrast, the Ni NPs formed in the absence of Pt were very large and irregularly shaped. This suggests that Pt atoms play an important role in facilitating the formation of cubic-shaped NPs. Another significant observation was that the concentration of Pt atoms deposited on the cubes was greater in the case of Ni–Pd–Pt than in Ni–Pt. This suggests that the presence of Pd atoms may render the corners and edges of the cube more active. Similarly, it should be noted that the reduction of Pt ions was delayed when oleylamine was introduced to the system. When the synthesis of Pt particles was attempted both in the presence and absence of oleylamine, the formation of Pt nanoparticles was observed at 120 °C in the oleylamine-free case. On the other hand, in the presence of oleylamine the Pt particles began to form at temperatures no higher than 170 °C. In contrast, the formation of Ni and Pd particles was not influenced by the presence of oleylamine. These experimental results partially explain the formation of the novel structure observed in this study. Further study will however be necessary to fully understand the mechanism of formation of this novel Pd-core–Ni-Pd shell in a Pt nanocage.
The catalytic potential of these particles was assessed by comparing conversion rates in the hydrogenation of 1-octene to n-octane. The results are illustrated in Fig. 6.
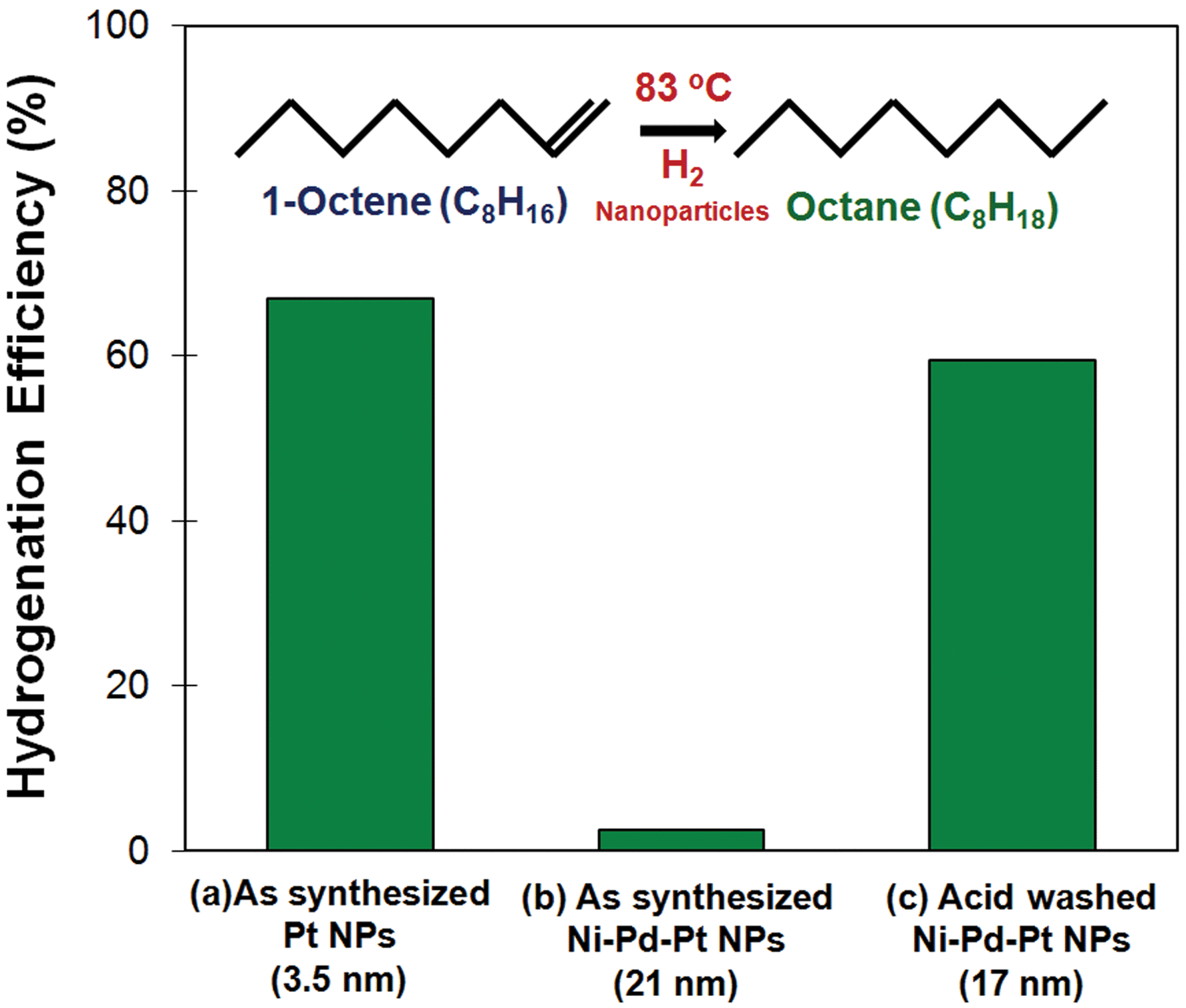 | ||
| Fig. 6 Conversion rates of the hydrogenation of 1-octene to n-octane, using a similar weight of metal catalyst, for (a) as-synthesized Pt (3.5 nm), (b) as-synthesized Ni–Pd–Pt (21 nm), and (c) acid-treated Ni–Pd–Pt (17 nm). | ||
It is seen that the conversion rate of 1-octene to n-octane in the presence of 3.5 nm Pt was very high when Ni–Pd–Pt particles were used in the as-synthesized state. On the other hand, their performance became comparable with Pt nanoparticles when the Ni–Pd–Pt particles were washed with acid. The reason for this was investigated and the presence of oleylamine confirmed in the as-prepared samples (ESI, Fig. S3 and S4†). However, washing the particles either with a mixture of toluene and methanol or with nitric acid helped to remove a considerable proportion of the oleylamine molecules and make them available for catalytic action. In addition, the removal of excess metallic nickel deposited on the surface of the cubes would also contribute to an improvement in the conversion rate. As shown in Fig. 1(c), when the reaction time is prolonged, the nickel ions remaining in solution begin to deposit on the surface of the cubes and inhibit the exposure of Pt atoms at the edges and corners of the cube.
We believe that the elucidation of the mechanism of formation of these novel nanostructures will allow us to control the proportion of Pt atoms deposited at the edges and corners of the cube, and consequently influence the catalytic or other functional properties of these particles. The elucidation of the mechanism of formation of bimetallic Ni–Pt particles by the authors is expected to be published shortly and it is anticipated that this will pave the way for the improved design of multifunctional Ni–Pd–Pt particles.
4. Conclusions
We have reported the synthesis of novel Pd-core–Ni-Pd-shell NPs in a Pt cage. Although the structural, morphological and atomic distributions of these NPs are understood, the factors that facilitate the formation of such an unusual nanostructure have still to be fully determined. Further investigation is expected to pave the way for the synthesis of multi-functional particles with novel structures. Experimental work to elucidate the mechanism of crystal formation and to explore the potential of these particles in a variety of fields is in hand.Acknowledgements
This study was supported by Grant-in Aid for Basic Research (B) and 22310064 and Challenging Exploratory Research 25600028 from the Ministry of Education, Science, Culture and Sport of Japan.References
- R. R. Barefoot and J. C. Van Loon, Anal. Chim. Acta, 1996, 334, 5 CrossRef CAS.
- J. Lipkowski and P. N. Ross, Electrocatalysis, Wiley-VCH, New York, 1998 Search PubMed.
- A. Serov and C. Kwak, Appl. Catal., B, 2009, 90, 313 CrossRef CAS PubMed.
- S. I. Lim, I. Ojea-Jimenez, M. Varon, E. Casals, J. Arbiol and V. Puntes, Nano Lett., 2010, 10, 964 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493 CrossRef CAS PubMed.
- X. Huang, Y. Li, Y. Li, H. Zhou, X. Duan and Y. Huang, Nano Lett., 2012, 12, 4265 CrossRef CAS PubMed.
- K. Lee, S. W. Kang, S.-U. Lee, K.-H. Park, Y. W. Lee and S. W. Han, ACS Appl. Mater. Interfaces, 2012, 4, 4208 CAS.
- C. Cui, L. Gan, H.-H. Li, S.-H. Yu, M. Heggen and P. Strasser, Nano Lett., 2012, 12, 5885 CrossRef CAS PubMed.
- L. Liu, G. Samjeske, S. Nakamatsu, O. Sekikawa, K. Nagasawa, S. Takao, Y. imaizumi, T. Yamamoto, T. Uruga and Y. Iwasawa, J. Phys. Chem. C, 2012, 116, 23453 CAS.
- J. Chen, B. Lim, E. P. Lee and Y. Xia, Nano Today, 2009, 4, 81 CrossRef CAS PubMed.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924 CAS.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663 CrossRef CAS PubMed.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097 CrossRef CAS PubMed.
- B. Jeyadevan, K. Urakawa, A. Hobo, N. Chinnasamy, K. Shinoda, K. Tohji, D. D. J. Djayaprawira, M. Tsunoda and M. Takahashi, Jpn. J. Appl. Phys., 2003, 42, L350 CrossRef CAS.
- C. N. Chinnasamy, B. Jeyadevan, K. Shinoda and K. Tohji, J. Appl. Phys., 2003, 93, 7583 CrossRef CAS PubMed.
- L. Jhon, C. Huaman, S. Fukao, K. Shinoda and B. Jeyadevan, CrystEngComm, 2011, 13, 3364 RSC.
- T. Itoh, M. Uebayashi, K. Tohji and B. Jeyadevan, Electrochemistry, 2010, 78, 157 CrossRef CAS PubMed.
- L. Jhon, C. Huaman, N. Hironaka, S. Tanaka, K. Shinoda, H. Miyamura and B. Jeyadevan, CrystEngComm, 2013, 15, 729 RSC.
- F. U. Hillebrecht, J. C. Fuggle, P. A. Bennett and Z. ZoLierek, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 2179 CrossRef CAS.
- Y. Wu, D. Wang, P. Zhao, Z. Niu, Q. Peng and Y. Li, Inorg. Chem., 2011, 50, 2046 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03091e |
| This journal is © The Royal Society of Chemistry 2014 |