
Carbon nanofibers containing Si nanoparticles and graphene-covered Ni for high performance anodes in Li ion batteries†
Abstract
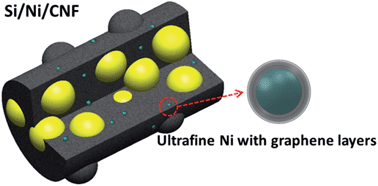
Freestanding, porous carbon nanofiber (CNF) composites containing Si nanoparticles and graphene-covered Ni particles are synthesized via one-pot electrospinning and thermal treatment. The electrodes made from the Si/Ni/CNF composites deliver a remarkable specific capacity of 1045 mA h g−1 at the 50th cycle at a current density of 100 mA g−1 and an excellent high rate capacity of 600 mA h g−1 at 1 A g−1 after 70 cycles with capacity retention of 81%. These values are among the best for similar electrospun Si-based CNF composite electrodes. Two major ameliorating mechanisms are responsible for the finding. The Si particles are fully encapsulated by the soft CNF matrix which serves as the stress buffer to relieve the volumetric stresses stemming from the intercalation/extraction of Li ions into Si and prevent re-agglomeration of Si particles during charge/discharge cycles. The amorphous carbon and the Ni particles surrounded by the crystallized graphene layers effectively form continuous conductive networks which in turn offer fast ion and electron transport paths, giving rise to high rate performance of the electrodes.
 Please wait while we load your content...
Please wait while we load your content...

