
Template-free solvothermal preparation of ZnO hollow microspheres covered with c planes†
Abstract
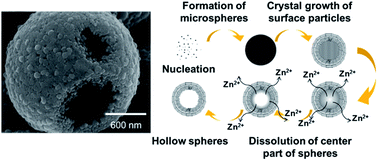
ZnO hollow microspheres were prepared by a solvothermal method, without using a template. The morphology and structure of the microspheres were investigated by scanning and transmission electron microscopies. A series of controlled experiments was carried out to better understand the formation mechanism. The sample prepared at 150 °C for 1 h consisted of ZnO microspheres 1–2 μm in diameter. Microspheres were composed of crystallites of tens of nm in diameter. The surfaces of the microspheres prepared at 150 °C for 3 h were surrounded by hexagonal pyramid-like crystals, of a few hundred nm in diameter. The sample prepared by reaction at 150 °C for 6 h consisted of hollow spheres, even in the absence of a template. The microsphere surface consisted of ZnO c planes. ZnO hollow microspheres prepared at 150 °C for 12 h exhibited intense narrow ultraviolet emission.
 Please wait while we load your content...
Please wait while we load your content...

