MnO2 nanoflakes anchored on reduced graphene oxide nanosheets as high performance anode materials for lithium-ion batteries†
Yong Cao,
Xionggui Lin,
Chenglong Zhang,
Cheng Yang,
Qian Zhang,
Weiqiang Hu,
Mingsen Zheng* and
Quanfeng Dong*
Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen, Fujian 361005, China. E-mail: qfdong@xmu.edu.cn; mszheng@xmu.edu.cn
First published on 24th June 2014
Abstract
A MnO2 nanoflake–reduced graphene oxide (MnO2–rGO) composite was synthesized by a facile solution method. The composite exhibited excellent electrochemical performance with a reversible capacity of 1430 mA h g−1 and 520 mA h g−1 at current densities of 0.1 A g−1 and 10 A g−1, respectively. MnO2 in the composite was proved to be fully activated and went through a complete conversion reaction. The improved kinetics in the MnO2–rGO composite electrode were evidenced by Electrochemical Impedance Spectroscopy (EIS) results, accounting for its extraordinary electrochemical properties compared with that in a simple MnO2–rGO mixture electrode. Due to the uniform dispersion and firm anchoring of MnO2 nanoflakes on the rGO surface, the volume expansion of MnO2 during the charge–discharge process was significantly alleviated. It showed excellent cyclic performance with an extremely large capacity of 1000 mA h g−1 maintained after 200 cycles at the current density of 1 A g−1.
Introduction
Lithium ion batteries (LIBs) have been dominant power sources for portable electronic devices since their commercialization in the 1990s and are also considered as the most promising large-scale energy storage devices for electrical vehicles and the green grid.1,2 To meet the demands of large-scale energy storage, higher energy density, good cycle stability, sound safety and lower cost are generally required in electrode materials related to both anodes and cathodes.3 Graphitic carbon, the most popularly used anode material for LIBs with the capacity of 372 mA h g−1 and low intercalation/deintercalation voltage (vs. Li/Li+), can deliver the theoretical energy density of ∼1370 W h kg−1, but suffers from its intrinsic safety problem of lithium dendrites when charged at high current density or overcharged.4 Li4Ti5O12, another anode material, exhibits excellent safety and cycle stability but is hindered by its low specific capacity (175 mA h g−1) and high working voltage (∼1.5 V vs. Li/Li+).5MnO2, one of transition metal oxides (TMOs), has been extensively investigated as the energy storage material for supercapacitors and LIBs, as well as the catalyst for Li–O2 batteries and water splitting, and so on, due to its low cost, environmental friendliness and natural abundance.6–8 In application as anode materials for large-scale LIBs, MnO2 has attracted tremendous attentions because of its high theoretical capacity up to 1232 mA h g−1 based on the conversion reaction denoted as: MnO2 + 4e− + 4Li+ → Mn + 2Li2O, which is about four times larger than that in commercialized graphite anodes. Moreover, its discharge voltage platform is as low as 0.4–0.5 V (vs. Li/Li+), indicating a higher energy density in contrast to other TMOs. However, it is still lack of merits as an electrode material because of its intrinsically low electrical conductivity and drastic capacity fading resulting from the large volume variation during the charge–discharge process. It is well accepted that the efficient way to improve the electrochemical performance of TMOs is to construct nanostructure composite with conductive materials, such as carbon tubes (CNTs), graphene, and so on.9–12 Among them, graphene is considered as an ideal carbon supporting owing to its superior electronic conductivity (106 S cm−1), high theoretical surface area (∼2630 m2 g−1) and good mechanical properties.13,14 Co2O3–graphene, SnO2–graphene, Mn3O4–graphene composites et al. have been demonstrated with the excellent electrochemical performances.15–19 On the other hand, the electrochemical properties of the composites were critically dependant on their structure configurations.20,21 A hierarchical sandwich-structured graphene–MnO2–graphene nanoribbons was reported as a stable LIBs anode by Li et al. while a nanoflaky MnO2–CNT composite electrode exhibiting a large reversible capacity of 801 mA h g−1 for the initial cycle without capacity fading within the first 20 cycles was reported by Xia et al.22,23 Despite of the successful fabrication of various MnO2–carbon nanocomposites with improved electrochemical performance, it is still difficult to meet the demands for the practical application of the MnO2 anodes due to their low specific capacity, poor cycle stability, and complicated synthesize strategy.
Herein, a facile strategy was developed to fabricate the MnO2–reduced graphene oxide nanosheets composite (MnO2–rGO composite) through the redox reaction of reduced graphene oxide and KMnO4 in an acid condition. MnO2 nanoflakes were in situ anchored on the rGO surface. Ascribing to its unique character of structure, the composite exhibited excellent electrochemical performance. It can deliver a reversible capacity of 1430 mA h g−1 at a current density of 0.1 A g−1. In particular, a capacity of 520 mA h g−1 can be achieved at the current density up to 10 A g−1. In addition, it also exhibited excellent cycle stability. No obvious capacity fading was found even after 200 cycles with a capacity of 1000 mA h g−1 at 1.0 A g−1. To our knowledge, it is the best result for MnO2 anodes to date.
Experimental section
Materials preparation
Graphene oxide (GO) was prepared by the modified Hummers' chemical methods and then thermally reduced into rGO at 1500 °C in N2 atmosphere (see ESI†). The MnO2–rGO composite was synthesized by the redox reaction of the rGO with KMnO4 in 0.5 M H2SO4 solution. The MnO2–rGO mixture was prepared by the direct balling mixture of pure MnO2 and rGO with the same mass ratio as that of the MnO2–rGO composite. The pure MnO2 was prepared with the same procedure as that of the MnO2–rGO composite but with excess amount of KMnO4.Materials characterizations
The crystallographic information were investigated by powder X-ray diffraction (XRD, Philips X'pert Pro Super X-ray diffractometer using Cu Kα radiation with λ = 1.5418 Å) at a range of 10–80. The detailed morphologies of the as prepared samples were characterized by field emission scanning electron microscopy (FESEM, HITACHI S-4800) and transmission electron microscopy (TEM, JEM-1400). A Pryis Diamond TG/DTA analyzer (Perkin Elmer) was employed to determine the mass content of MnO2 in the composite under a N2/O2 atmosphere at a heating rate of 5 °C min−1.Electrochemical measurements
80 wt% of the active material, 10 wt% acetylene black and 10 wt% water soluble polymer n-lauryl acrylate (LA, Chengdu, China) were mixed and mechanically milled as the working electrodes. The slurries were coated onto Cu foils and dried at 60 °C under vacuum overnight. Then, Coin cells (CR2016) were assembled in an Argon-filled glove box with lithium foil as the counter electrode, Cellgard2400 as separator and 1 M LiPF6 in ethylene carbonate, dimethyl carbonate and diethyl carbonate (EC–DMC–DEC, v/v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) as the electrolyte. The cycling performance and rate tests were carried out on a NEWARE BTS-5 V/5 mA type charger (Shenzhen, China) within the voltage range of 0.02–3.0 V. The specific capacity was calculated on the basis of the weight of MnO2 but for some specific instructions. All experiments were conducted at room temperature.
:1:1) as the electrolyte. The cycling performance and rate tests were carried out on a NEWARE BTS-5 V/5 mA type charger (Shenzhen, China) within the voltage range of 0.02–3.0 V. The specific capacity was calculated on the basis of the weight of MnO2 but for some specific instructions. All experiments were conducted at room temperature.
Results and discussion
XRD and TGA analysis
Fig. 1a shows the XRD patterns of as-prepared rGO nanosheets, pure MnO2 and MnO2–rGO composite. A solely broad diffraction peak at ∼25° was observed in the pattern of rGO nanosheets, corresponding to the average d-spacing of graphene layer. The same four diffraction peaks can be observed in both of the patterns in pure MnO2 and MnO2–rGO nanocomposite, which are indexed well to birnessite-type MnO2 (JCPDS card no. 80-1098). Noted, the diffraction peak related to the rGO nanosheets cannot be observed in the pattern of MnO2–rGO composite, indicating the homogenous coating of MnO2 nanoflakes on the rGO surface suppressing the stacking of graphene layers.24 The mass percentage of MnO2 in the composite was measured by the Thermal Gravimetric Analysis (TGA) method as shown in the Fig. 1b with the temperature ranging from room temperature to 750 °C. The TGA profile demonstrates two main weight loss regions. The first weight loss of about 6.5% below 250 °C is commonly corresponding to the interlayer water in the composite while the second weight loss after 300 °C can be contributed to the oxidation of rGO. The mass percentage of MnO2 is calculated to be ∼78%. | ||
| Fig. 1 (a) X-ray diffraction patterns of the as prepared rGO nanosheets, pure MnO2 and MnO2–rGO composite. (b) TGA profile of the MnO2–rGO composite. (c) The SEM image of the rGO nanosheets. (d) The SEM and (e) TEM images of the MnO2–rGO composite. (f) The SEM and (g) TEM images of the pure MnO2. | ||
Morphological and structural characterization
The morphology characterization of the as-prepared three samples was investigated by scanning electron microscopy (SEM) and Transmission electron microscopy (TEM). The near-transparent rGO nanosheet with wrinkled surface structure is presented in Fig. 1c. The highly dispersive rGO nanosheets act as a 3D conductive matrix and form interconnected micro- and meso-pores, which would definitely facilitate the electron transport and the diffusion of Li ions. Fig. 1d shows the homogeneous growth of MnO2 nanoflakes on the surface of rGO nanosheets with rGO nanosheets maintaining the highly dispersive status. Similarly, the TEM image of the MnO2–rGO composite as seen in Fig. 1e further evidences the uniform growth of MnO2 nanoflakes on rGO surface. The bulk MnO2 microflower assembled from its nanoflakes is morphologically displayed in the SEM image of Fig. 1f and TEM image of Fig. 1g. Fig. 2a–d display the TEM EDX mapping of the MnO2–rGO composite which clearly show the uniform elemental distribution of C, Mn and O in the composite. HRTEM image in Fig. 2e reveals the measured interplanar spacing of birnessite-type MnO2 of about 0.67 nm. Apparently, the anchoring of MnO2 nanoflakes on the rGO surface can effectively suppress the stacking of graphene layers. Nearly every MnO2 nanoflake is securely “bonded” with the rGO matrix, and thus, electrons can transfer from the rGO to every MnO2 flake timely, which lays the basis for excellent electrochemical performance of the MnO2–rGO composite. Understandably, this effect is absent in the only bulk MnO2 microflowers and the simple MnO2–rGO mixture electrode. | ||
| Fig. 2 (a)–(d) TEM EDX mapping of the MnO2–rGO composite. (e) HRTEM image of the MnO2 nanoflake. | ||
Electrochemical performance
The electrochemical performances of the as-prepared MnO2–rGO composite and MnO2–rGO mixture were investigated by galvanostatic charge–discharge method. Fig. 3a compares the 1st and 2nd charging–discharge profiles of the MnO2–rGO composite with those in the MnO2–rGO mixture electrode at a current density of 0.2 A g−1 within the voltage region between 0.02 V and 3.0 V. A high reversible capacity up to 1100 mA h g−1 (∼1320 mA h g−1 calculated on the weight of MnO2) was characterized in MnO2–rGO composite anode in the first cycle, which is even slightly higher than its theoretical capacity of 1233 mA h g−1 upon the conversion reaction of MnO2 + 4Li+ + 4e− → 2Li2O + Mn. The exceeding capacity is probably coming from the lithium ion adsorption/desorption on the Li2O/Mn matrix.25 The initial columbic efficiency is 63.1%, which is not higher than other MnO2 anodes reported, mainly resulting from the SEI film formation, electrolyte decomposition, and irreversible reaction of the electrode.23,26 As a comparison, the 1st and 2nd discharge–charge curves of the MnO2–rGO mixture electrode are also shown in Fig. 3a. The MnO2–rGO mixture electrode also can deliver a large initial discharge capacity of ∼1600 mA h g−1 with the first discharge curve similar as that of the MnO2–rGO composite anode, implying a similar initial discharging process. But only a reversible capacity of ∼600 mA h g−1 was delivered in the MnO2–rGO mixture electrode in the first cycle with the columbic efficiency down to ∼38%. There is quite difference in the 1st charge processes between the two samples. Two obvious different electrochemical processes could be observed in the MnO2–rGO composite electrode. One is from 0.02 V to 1.5 V, the other is from 1.5 V to 3.0 V. The capacities of the two processes are both ∼580 mA h g−1, indicating that the anodic oxidation process of Mn and Li2O might possess two steps: Mn + Li2O → 2Li+ + 2e− + MnO, and MnO + Li2O → 2Li+ + 2e− + MnO2. In contrast, only one plateau around 1.1 V is observed in the charge profile of the MnO2–rGO mixture with the capacity of ∼600 mA h g−1, which is almost half of the theoretical value, corresponding to a two-electron transfer process. It implies that MnO is the possible anodic oxidation reaction product for the MnO2–rGO mixture electrode. In addition, the voltage platform at ∼0.5 V can both be observed in the 2nd discharge process in both two samples, which is also similar as the initial discharge process. As mentioned above, MnO is the electrochemical oxidation product of MnO2–rGO mixture in the charge process, which is further evidenced by the comparable 2nd discharge profile of MnO–rGO mixture with that of MnO electrodes in the literatures.27 Furthermore, the capacity of MnO2–rGO composite in the plateau is about half of its theoretical value. It demonstrates that the reduction process of MnO2–rGO composite may also possess two steps: MnO2 + 2Li+ + 2e− → Li2O + MnO, and MnO + 2Li+ + 2e− → Li2O + Mn. The result of the cyclic voltammetry test shown in Fig. S2 (ESI†) also clearly reveals two couples of oxidation/reduction peaks in the MnO2–rGO composite electrode and one couple in the MnO2–rGO mixture anode, which are consistent with the charge–discharge curves presented above. | ||
| Fig. 3 (a) A contrast of the 1st and 2nd charge–discharge voltage profiles of the MnO2–rGO composite and the MnO2–rGO mixture electrodes. (b) Mn 2p XPS spectra in the charged MnO2–rGO composite and the MnO2–rGO mixture electrodes, respectively. (c) Nyquist plots of the MnO2–rGO composite and the MnO2–rGO mixture. The insert table lists the detailed impedance parameters derived by the equivalent circuit model shown in (Fig. 2d). | ||
To further determine the oxidation state of the charging products, XPS and a chromogenic reaction test were performed. Fig. 3c shows the fitted Mn 2p XPS spectrums of the MnO2–rGO composite and the MnO2–rGO mixture electrodes at the state of full-charged. The spin energy of Mn 2p3/2 and Mn 2p1/2 of the charged MnO2–rGO composite electrode are centered at 641.7 and 653.5 eV, respectively. The energy separation is calculated to be 11.8 eV, which is in a good agreement with that of MnO2.28 This soundly evidences that MnO2 is the final product of charged MnO2–rGO composite electrode, which confirms the results of the electrochemical measurement. As comparison, the peaks of Mn 2p3/2 and Mn 2p1/2 in the charged MnO2–rGO mixture electrode are very weak and centered at 641 and 653 eV, which well agree with those reported characteristic binding energies in MnO.28 The chromogenic reaction test shown in Fig. S4 (ESI†) also demonstrates the similar results as above. So we can conclude that the MnO2 in the composite went through a complete conversion reaction other than that in the mixture. The pictures of the charged electrodes shown in Fig. S4 (ESI†) also clearly depict the severe peeling of the active material from the current collector in the MnO2–rGO mixture electrode. But that is alleviated in the MnO2–rGO composite electrode.
Fig. 3c shows the results of the electrochemical impedance spectra of the MnO2–rGO composite and the MnO2–rGO mixture electrodes. The semicircle in the high and middle frequency range are corresponding to the resistance of SEI film (RSEI) and charge transfer (RCT), respectively, while the sloping line in the lower frequency represents the Warburg impedance (Zw) of Li-ion diffusion. The impedance parameters are listed in the inset table in Fig. 3c, derived by the equivalent circuit model shown in Fig. 3d. The RSEI and RCT of the MnO2–rGO composite are 2.631 and 9.747 Ω, which is about one magnitude lower than those of the MnO2–rGO mixture. It means that the charge transfer kinetic in the MnO2–rGO composite is greatly improved, resulting from the firmly combination of MnO2 nanoflakes with the excellently conductive rGO.
The rate capability and cycle performance of MnO2–rGO composite electrode and MnO2–rGO mixture electrode were also evaluated contrastively. As shown in Fig. 4, the MnO2–rGO composite exhibits not only significantly smaller irreversible capacity during the 1st cycle, but also extremely larger capacities at various current densities. The initial reversible capacity of the MnO2–rGO composite electrode at 0.1 A g−1 is as high as 1430 mA h g−1 and it can be maintained at 1000 mA h g−1 at 2 A g−1. The corresponding values of the MnO2–rGO mixture electrode are only 600 mA h g−1 and 95 mA h g−1, respectively. Remarkably, the MnO2–rGO composite electrode can still deliver a large capacity of ∼520 mA h g−1 at a high current density of 10 A g−1 and then recover to ∼1400 mA h g−1 when the current density is switched back to 0.2 A g−1 after 35 cycles after different current densities imposed. The charge–discharge voltage profiles of the MnO2–rGO composite electrode at different current densities are shown in Fig. S3.† Under every current density, two steps can be observed in both discharging and charging processes.
 | ||
| Fig. 4 Rate performances of the MnO2–rGO composite and the MnO2–rGO mixture electrodes at different current densities with the voltage window between 0.02 V and 3 V. | ||
Fig. 5a shows the capacities of the MnO2–rGO composite and the MnO2–rGO mixture at the current density of 0.2 A g−1 within the initial 50 cycles. Except for the first two cycles, no obvious fading is observed in the MnO2–rGO composite electrode with the capacity keeping at ∼1350 mA h g−1. In contrast, the capacity of the MnO2–rGO mixture fades rapidly from 1400 mA h g−1 to 200 mA h g−1 after about 10 cycles. The MnO2–rGO composite electrode also exhibited stable cycling performance at a high current density of 1 A g−1, as displayed in Fig. 5b. The cell retained nearly 100% of its initial capacity even after prolonged cycling over 200 cycles, even exhibiting a slightly capacity increase in the first 80 cycles. The coulombic efficiency of the whole cycling process was above 99%. What is even more exciting is that the rate procedure (from 0.2 A g−1 to 10 A g−1) could be stably repeated for extended 2 times without capacity decay, as shown in Fig. 5c. The excellent rate capability and cyclic performances indicate improved reaction kinetics and stability of the composite electrode.
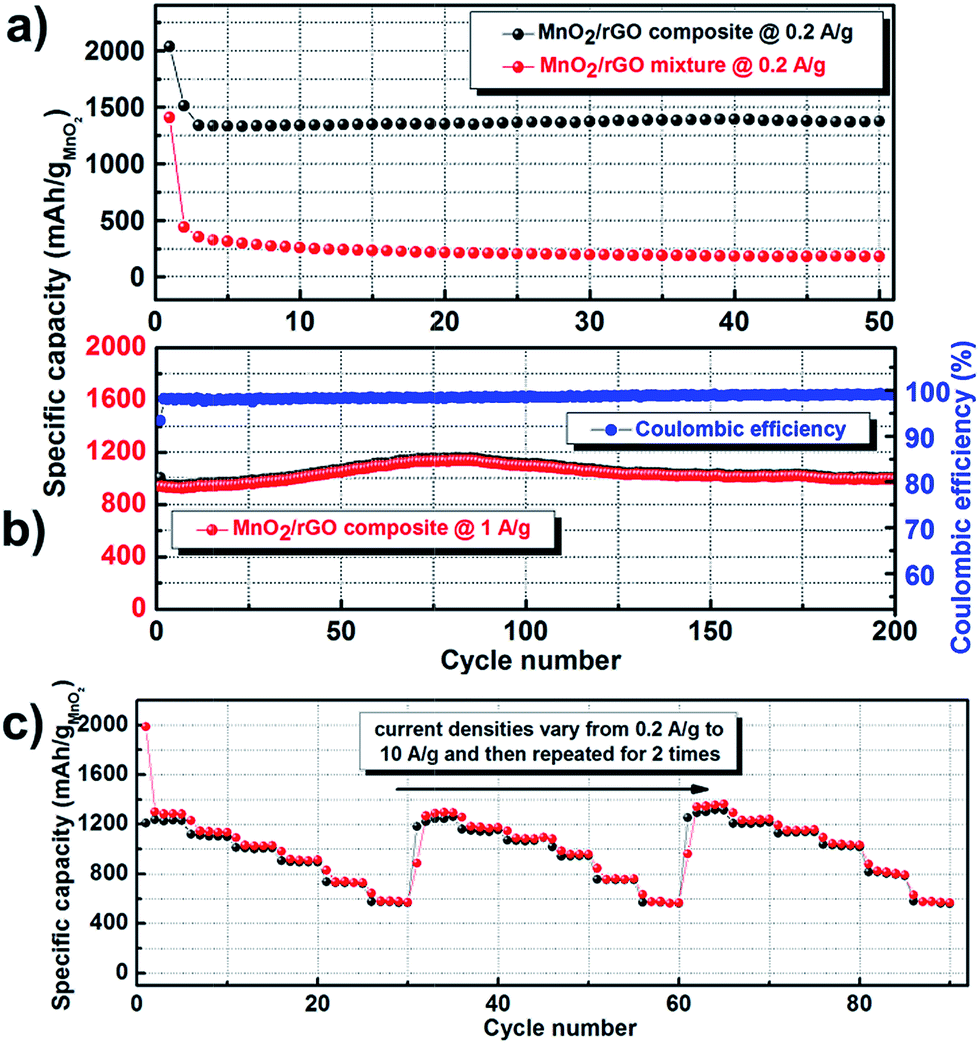 | ||
| Fig. 5 (a) A comparison between the cycling performance of the MnO2–rGO composite and (b) Cycling performance and coulombic efficiency of the MnO2–rGO composite electrode at 1 A g−1. (c) Cycling performance of the MnO2–rGO composite electrode with an extended repeat of the rate procedure (from 0.2 A g−1 to 10 A g−1) for 2 times. | ||
Conclusion
In summary, a MnO2–rGO composite was synthesized as a high performance anode for LIBs. The composite exhibited extremely high rate capability with 1430 mA h g−1 and 520 mA h g−1 at the current density of 0.1 A g−1 and 10 A g−1, respectively, due to the fast kinetic character for both electron and lithium ion transfer resulting from its particular structure of MnO2 nanoflakes anchoring on conductive rGO nanosheets. The composite also exhibited the excellent cyclic stability owing to the highly dispersive structure which can accommodate the volume change of MnO2 during repeated cycles. It is a promising anode for high energy density LIBs and presents potential applications in large-scale energy storage systems.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638–654 CAS.
- K.-S. Park, A. Benayad, D.-J. Kang and S.-G. Doo, J. Am. Chem. Soc., 2008, 130, 14930–14931 CrossRef CAS PubMed.
- Y. Cao, Z. Wei, J. He, J. Zang, Q. Zhang, M. Zheng and Q. Dong, Energy Environ. Sci., 2012, 5, 9765–9768 CAS.
- Y. Qin, J. Lu, P. Du, Z. Chen, Y. Ren, T. Wu, J. T. Miller, J. Wen, D. J. Miller, Z. Zhang and K. Amine, Energy Environ. Sci., 2013, 6, 519–531 CAS.
- T. Zhai, F. Wang, M. Yu, S. Xie, C. Liang, C. Li, F. Xiao, R. Tang, Q. Wu, X. Lu and Y. Tong, Nanoscale, 2013, 5, 6790–6796 RSC.
- H. Chang and H. Wu, Energy Environ. Sci., 2013, 6, 3483–3507 CAS.
- Y. Li, Q. Zhang, J. Zhu, X.-L. Wei and P. K. Shen, J. Mater. Chem. A, 2014, 2, 3163–3168 CAS.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2008, 9, 72–75 CrossRef PubMed.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Müllen, ChemSusChem, 2010, 3, 236–239 CrossRef CAS PubMed.
- H. Wang, L.-F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- X. Zhou, L.-J. Wan and Y.-G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS PubMed.
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 CAS.
- L. Li, A.-R. O. Raji and J. M. Tour, Adv. Mater., 2013, 25, 6298–6302 CrossRef CAS PubMed.
- H. Xia, M. Lai and L. Lu, J. Mater. Chem., 2010, 20, 6896–6902 RSC.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- J. Jamnik and J. Maier, Phys. Chem. Chem. Phys., 2003, 5, 5215–5220 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- J. Zang, H. Qian, Z. Wei, Y. Cao, M. Zheng and Q. Dong, Electrochim. Acta, 2014, 118, 112–117 CrossRef CAS PubMed.
- Y. Xia, Z. Xiao, X. Dou, H. Huang, X. Lu, R. Yan, Y. Gan, W. Zhu, J. Tu, W. Zhang and X. Tao, ACS Nano, 2013, 7, 7083–7092 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02838d |
| This journal is © The Royal Society of Chemistry 2014 |