Nanolamellar triblock of poly-D,L-lactide–δ-valerolactone–D,L-lactide with tuneable glass transition temperature and crystallinity for use as a drug-delivery vesicle†
Nibedita Kasyapia and
Anil K. Bhowmick*ab
aDepartment of Materials Science and Engineering, School of Engineering and Technology, Indian Institute of Technology Patna, Patna 800013, India. E-mail: anilkb@rtc.iitkgp.ernet.in; director@iitp.ac.in; Fax: +91 612 2277384; Tel: +91 612 2552001
bDepartment of Chemistry, School of Basic Sciences, Indian Institute of Technology Patna, Patna 800013, India
First published on 2nd June 2014
Abstract
A biodegradable triblock copolymer, poly-D,L-lactide–δ-valerolactone–D,L-lactide, was synthesized by the ring-opening polymerization of δ-valerolactone and the sequential addition of the D,L-lactide monomer to the hydroxyl end of a functionalized poly-δ-valerolactone macroinitiator. The copolymer was then evaluated for its suitability as a drug-delivery vesicle. The effect of the monomer ratio, catalyst and initiator concentration on the structure was investigated using 1H-NMR, 13C-NMR and Fourier transform infrared spectroscopy, and gel-permeation chromatography. 1H-NMR confirmed the presence of the D,L-lactide segment as the terminal segment and δ-valerolactone as the mid-segment. 13C-NMR was used to study the block sequencing and extent of trans-esterification. The crystallization of the triblock was retarded compared with the pure poly-δ-valerolactone homopolymer due to the incorporation of the D,L-lactide moiety at the chain end of the poly-δ-valerolactone segment. The glass transition temperatures of the two blocks shifted depending on the ratio of the two monomers. The triblock, with molecular weights between 5000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, showed a separated morphology in the nanophase, with alternating stripes of amorphous and crystalline segments (3–6 nm) under transmission electron microscopy. The triblock was then fabricated into microspheres with an average diameter of 17.2 μm and was used to encapsulate salicylic acid. The formation of pores on the surface of the microsphere facilitated the release of the salicylic acid. The release profile displayed the characteristics of a potential carrier.
000 Da, showed a separated morphology in the nanophase, with alternating stripes of amorphous and crystalline segments (3–6 nm) under transmission electron microscopy. The triblock was then fabricated into microspheres with an average diameter of 17.2 μm and was used to encapsulate salicylic acid. The formation of pores on the surface of the microsphere facilitated the release of the salicylic acid. The release profile displayed the characteristics of a potential carrier.
Introduction
Increasing awareness of the need to protect the health of the global population, along with the depletion of fossil fuel reserves and the non-biodegradability of commodity plastics, has encouraged research into the potential applications of biomaterials due to their wide availability and non-toxic residues. As a consequence, novel strategies to increase the use of biodegradable polymers in everyday life, with the aim of solving the problem of time-resistant polymeric waste, have attracted much attention. Aliphatic polyesters derived from lactones are well recognized as biodegradable polymers. Among these, polylactide is the most widely studied biomaterial; there is a huge demand for this material in the biomedical industry.1 However, the limitations of polylactide2 have encouraged researchers to investigate other polyesters such as poly-ε-caprolactone and polyethylene glycol. Copolymerization with complementary monomers allows physical properties such as strength and dimensional stability to be tailored by adjusting the ratio of the individual blocks and by adding new blocks. We have previously reported the structure–property relationships of several copolymers.3,4 The ring-opening polymerization technique has been successfully used to synthesize copolymers of lactones such as glycolide, lactide (LA), β-butyrolactone, ε-caprolactone and 1,5-dioxepan-2-one.5–10 The architecture of the copolymers was tailored to yield random, diblock, triblock, graft and star copolymers by controlling the sequence of addition and varying the initiator catalyst system.11There have been preliminary reports of the homopolymerization of δ-valerolactone. Ring-opening coordination insertion polymerization and living cationic polymerization have been used efficiently for δ-valerolactone.12–14 However, a major hurdle is the cytotoxicity of the catalyst residues, which limits the commercialization of these processes. Among the catalysts, tin octoate has been approved by the US Food and Drug Administration15 and this catalyst is therefore preferred. The copolymerization of δ-valerolactone with other lactone monomers and characterization of the polymers derived from the respective monomers has also been carried out;16,17 however, there have been few reports of copolymer synthesis using δ-valerolactone and lactide.18 Lactide can be easily derived from corn starch and δ-valerolactone has been obtained as a by-product of the synthesis of adipic acid from tetrahydrofuran (THF).19 Characteristics such as the glass transition temperature, crystallinity and the morphology of the homopolymers can be tailored using the copolymerization technique. The monomer ratio in the synthesized copolymer may efficiently control the degradation rate of the copolymer. This new copolymer system can be successfully used as a drug-delivery system due to its biodegradable and biocompatible nature.
The state-of-the-art applications of biodegradable and biocompatible polymers are in the preparation of drug-delivery vesicles, biomimetic implants and the design of scaffolds for tissue engineering. Another improvised system used for drug delivery is the use of microspheres which can encapsulate the therapeutic agent. Copolymers from poly-ε-caprolactone or polyethylene glycol (PEG) in combination with glycolic acid and lactic acid are promising candidates for use as microspheres.20–25 Several papers have been published on a PEG system for micelle-assisted drug delivery.26,27 However, the crafting of microspheres using a triblock copolymer of δ-valerolactone and D,L-lactide has not yet been established.
The study reported here consisted of two parts. First, the stepwise synthesis of the new triblock copolymer, D,L-lactide–δ-valerolactone–D,L-lactide was carried out. Although the copolymerization technique is well established, we manipulated the synthesis with respect to the catalyst, initiator, time and temperature; the reactivity of the co-monomers is governed by these parameters. The triblock architecture of the copolymer is controlled by the sequential addition of the monomers. Organic solvents were not used during the preparation process, in order to make the method more environmentally friendly. The chemical composition was characterized using 1H-NMR, 13C-NMR and Fourier transform infrared (FTIR) spectroscopy. The crystallinity of the polymer was studied by wide-angle X-ray diffraction (WAXD) in powder form. In addition, gel-permeation chromatography (GPC) was used to determine the molecular weights and their distribution. The thermal properties were evaluated using differential scanning calorimetry (DSC). The morphology of the block copolymer was thoroughly investigated using transmission electron microscopy (TEM). A detailed investigation of the properties is essential in understanding the performance of the resulting polymer. This work has identified several factors which can be used to tailor the molecular weight, crystallinity and thermal properties of the copolymer which have not previously been reported for similar triblock polymers. The catalyst and initiator concentrations were varied to study their effect on the copolymerization reaction. Little work has so far been published on morphological studies of biodegradable block copolymers.28,29 The second part of the study involved the successful preparation of microspheres from the copolymer, the incorporation of salicylic acid into the microspheres and a study of the release of salicylic acid from the microspheres into phosphate-buffered saline (PBS) at pH 7.4. Field emission scanning electron microscopy (FESEM) was used to establish the formation of the microspheres and UV-visible spectroscopy was used to study the release of salicylic acid.
Materials and methods
D,L-Lactide was used as received and was stored and handled under a nitrogen atmosphere in a glove box. δ-Valerolactone and tin(II) 2-ethyl hexanoate were distilled under reduced pressure and stored under a nitrogen environment. 1,6-Hexanediol was used as received and stored under vacuum. All the chemicals were purchased from Sigma-Aldrich, Germany. All other solvents were used as received without further purification.1H-NMR spectra were recorded using a Model AVANCE III 400 Ascend Bruker spectrometer operating at 400 MHz; the 13C-NMR data were collected at 100 and 400 MHz. The samples for NMR were prepared in CDCl3 solution and the chemical shifts were reported as δ values (ppm) relative to the 1H signals from the protic solvent (7.26 ppm for CDCl3). The molecular weight distribution of the homopolymers and copolymers was measured using an Agilent PL-GPC 50 Integrated System with a PLgel 5 μm Mixed-D column equipped with a refractive index detector using THF as a solvent. The FTIR spectra were recorded on KBr pellets using Perkin-Elmer Spectrum 400 machine in the spectral range 4000–530 cm−1, with a total of 16 scans per sample. WAXRD analysis was carried out using a Rigaku TT RAX 3XRD machine with CuKα (0.154 nm) as the radiation source at 50 kV. DSC was conducted in a Perkin-Elmer DSC8000 calorimeter under a nitrogen atmosphere. Samples in hermetically sealed aluminum pans were analyzed as follows: the samples were equilibrated at 80 °C at 10 °C min−1 then cooled to −80 °C at 10 °C min−1, followed by reheating to 80 °C at 2 °C min−1. The glass transition temperature was recorded in the second heating cycle. The morphological studies were executed using a Tecnai G2 transmission electron microscope at 120 kV in the transmission mode. The samples were prepared by embedding into epoxy resin; the region of interest was sectioned by a diamond knife of 100 nm thickness. The sections were stained with OsO4 for 4 h at 72 °C, followed by staining with freshly prepared RuO4 for 4 h.
Synthesis of poly-δ-valerolactone
A typical procedure for synthesizing poly-δ-valerolactone is reported here. δ-Valerolactone (26.97 g, 0.2694 mol) and 1,6-hexanediol (31.8 mg, 0.2694 mmol) were added to a 25 mL round-bottomed flask equipped with a Teflon-coated magnetic stirrer under a nitrogen atmosphere. The sealed reaction vessel was then placed in an oil-bath and heated to 90 °C at a heating rate of 2 °C min−1, followed by the addition of Sn(Oct2) (0.109 g, 0.2694 mmol). The reaction mixture was then heated to 120 °C for 48 h. After cooling, the solid reaction product was dissolved in dichloromethane and precipitated in excess methanol. The solvent was removed under reduced pressure at room temperature for 2 days. The sample was designated sample D1. The time of the reaction was varied from 12 to 48 h. The ratio of the monomer (M) to initiator (I) concentration, [M]0/[I]0, was varied from 333 to 2000, while the ratio of the monomer (M) to catalyst (cat), [M]0/[cat]0, ranged between 333 and 2000. The reaction parameters for synthesizing D0, D1, D2, D3 and D4 are summarized in Table 1. The product was characterized by FTIR, 1H-NMR, 13C-NMR and size-extrusion chromatography (SEC). 1H-NMR (400 MHz, CDCl3, δ): 7.3, 4.1, 3.7, 2.4, 1.7. 13C-NMR (400 MHz, CDCl3, δ): 173.5, 171.6, 69.7, 68.1, 64.1, 33.9, 30.0, 28.2, 25.8, 22.5, 21.6, 19.3. The poly-D,L-lactide homopolymer was also synthesized (see ESI† for details) and characterized by the above techniques. 1H-NMR (400 MHz, CDCl3, δ): 7.3, 5.2, 3.8, 2.4, 1.6. 13C-NMR (400 MHz, CDCl3, δ): 169.6, 72.5, 69.4, 66.7, 20.5, 16.7, 15.8.| Conditions | wt% ratio | Catalyst | Initiator | Remarks | |
|---|---|---|---|---|---|
| D0 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.3 mol% catalyst, 0.3 mol% initiator | — | Tin octoate | 1,6-Hexanediol | |
| D1 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.1 mol% catalyst, 0.1 mol% initiator | — | Tin octoate | 1,6-Hexanediol | |
| D2 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.05 mol% catalyst, 0.05 mol% initiator | — | Tin octoate | 1,6-Hexanediol | |
| D3 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.1 mol% initiator, 0.05 mol% catalyst | — | Tin octoate | 1,6-Hexanediol | |
| D4 | Polymerization of δ-valerolactone at 120 °C for 12 h + 0.126 mol% initiator, 0.1 mol% catalyst | — | Tin octoate | 1,6-Hexanediol | |
| DL15050 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.1 mol% catalyst, 0.1 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.05 mol% catalyst | 50:50 |
Tin octoate | 1,6-Hexanediol | Variable composition taking D1 as macroinitiator |
| DL16634 | Polymerization of δ valerolactone at 120 °C for 48 h + 0.1 mol% catalyst, 0.1 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.05 mol% catalyst | 66:34 |
Tin octoate | 1,6-Hexanediol | |
| DL18020 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.1 mol% catalyst, 0.1 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.05 mol% catalyst | 80:20 |
Tin octoate | 1,6-Hexanediol | |
| DL25050 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.05 mol% catalyst, 0.05 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.05 mol% catalyst | 50:50 |
Tin octoate | 1,6-Hexanediol | Variable composition taking D2 as macroinitiator |
| DL26634 | Polymerization of delta valerolactone at 120 °C for 48 h + 0.05 mol% catalyst, 0.05 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.05 mol% catalyst | 66:34 |
Tin octoate | 1,6-Hexanediol | |
| DL28020 | Polymerization of delta valerolactone at 120 °C for 48 h + 0.05 mol% catalyst, 0.05 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.05 mol% catalyst | 80:20 |
Tin octoate | 1,6-Hexanediol | |
| D2L1 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.05 mol% catalyst, 0.05 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.02 mol% catalyst | 66:34 |
Tin octoate | 1,6-Hexanediol | |
| D2L2 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.05 mol% catalyst, 0.05 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.05 mol% catalyst | 66:34 |
Tin octoate | 1,6-Hexanediol | |
| D2L3 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.05 mol% catalyst, 0.05 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.126 mol% catalyst | 66:34 |
Tin octoate | 1,6-Hexanediol | |
| DL4060 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.3 mol% catalyst, 0.3 mol% initiator, addition of D,L-lactide at 140 °C for 6 h + 0.126 mol% catalyst | 40:60 |
Tin octoate | 1,6-Hexanediol | Variable composition taking D0 as macroinitiator |
| DL6040 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.3 mol% catalyst, 0.3 mol% initiator addition of D,L-lactide at 140 °C for 6 h + 0.126 mol% catalyst | 60:40 |
Tin octoate | 1,6-Hexanediol | |
| DL7030 | Polymerization of δ-valerolactone at 120 °C for 48 h + 0.3 mol% catalyst, 0.3 mol% initiator addition of D,L-lactide at 140 °C for 6 h + 0.126 mol% catalyst | 70:30 |
Tin octoate | 1,6-Hexanediol |
Synthesis of D,L-lactide–δ-valerolactone–D,L-lactide triblock copolymer
In the glove box, D1 (3.16 g, 31.56 mmol) and D,L-lactide (3.17 g, 21.99 mmol) were added to a 25 mL round-bottomed flask equipped with a Teflon-coated magnetic stirrer under a nitrogen atmosphere. The sealed reaction vessel was then placed in an oil-bath and heated to 90 °C, followed by the addition of Sn(Oct2) (0.01084 g, 0.0268 mmol). The reaction mixture was then heated to 140 °C for 8 h. The reaction product was then cooled to room temperature and dissolved in dichloromethane followed by precipitation in excess methanol. The recovered solid was dried under reduced pressure for 2 days. The sample was marked as DL15050. The resulting copolymer was thoroughly characterized by FTIR, 1H-NMR, 13C-NMR and GPC. Several copolymers with variable feed compositions were prepared using the homopolymer as the macroinitiator with synthesis at different [M]0/[I]0 and [M]0/[cat]0 ratios. The sample details are given in Table 1. 1H-NMR (400 MHz, CDCl3, δ): 7.3, 5.2, 4.1, 3.7, 2.3, 1.7, 1.6, 1.3. 13C-NMR (400 MHz, CDCl3, δ): 173.3, 169.6, 169.4, 169.1, 69.4, 69.2, 68.9, 68.3, 66.7, 65.0, 63.9, 33.7, 33.4, 33.1, 28.1, 27.9, 27.7, 21.4, 21.2, 20.5, 16.7, 16.6, 15.8.Preparation of microspheres containing salicylic acid from D,L-lactide–δ-valerolactone–D,L-lactide triblock copolymer
Microspheres loaded with salicylic acid were prepared by a solvent evaporation technique in an oil-in-water emulsion. First, both salicylic acid (200 mg) and the D,L-lactide–δ-valerolactone–D,L-lactide triblock copolymer (400 mg) were dissolved in 10 mL of dichloromethane and sonicated for 15 minutes. The solution was then added dropwise to an aqueous medium containing 0.5 wt% Tween 20 (polyethylene glycol sorbitan monolaurate) while mixing vigorously at 1000 rpm using a mechanical stirrer. The stirring was continued for 1 h to evaporate the dichloromethane and the microspheres were collected by centrifugation at 12000 rpm. The microspheres loaded with salicylic acid were thoroughly washed with water and dried in a vacuum oven for 2 days.
In vitro release study of salicylic acid from loaded triblock copolymeric microspheres
The in vitro release study was performed in pH 7.4 PBS. A 0.65 mg mass of microspheres loaded with salicylic acid was dispersed in 100 mL of PBS buffer. The release medium was placed in an orbital shaker at 160 ± 10 rpm and an aliquot of 0.5 mL, taken out from the medium at set time intervals for analysis, was replaced by an equal volume of fresh buffer solution. The concentration of salicylic acid was measured using a Shimadzu UV-visible spectrophotometer (Model UV-2550). Salicylic acid showed a strong absorption band at 294 nm associated with a weak absorption at 229 nm. The absorbance of the PBS buffer was negligible in this region. The concentration of the salicylic acid in the release medium was relatively dilute and hence the solution was assayed spectrophotometrically at 294 nm, the stronger absorption band, at predetermined time intervals.The microspheres were examined under FESEM (Hitachi S-4800 microscope) at an accelerating voltage of 10 kV. Samples of the microspheres before and after release were prepared by dropping a microparticle suspension onto double-sided carbon adhesive tape adhered on the stub and coated with platinum using a Hitachi E-1010 ion sputter system. FTIR spectroscopy in the KBr mode was used to confirm the incorporation of salicylic acid into the microspheres. Thermogravimetric analysis was carried out on a TA instrument SDT Q600 system at a ramp rate of 10 °C min−1 under a nitrogen atmosphere from room temperature to 500 °C.
Results and discussion
Synthesis and characterization of poly-δ-valerolactone
A bulk ring-opening polymerization technique was adopted for the synthesis of the poly-δ-valerolactone homopolymer using tin 2-ethyl hexanoate as the catalyst and 1,6-hexanediol as the initiator at 120 °C for 48 h. A series of reactions was investigated by varying the monomer to initiator ratio from 333 to 2000 while the catalyst to monomer ratio was also varied in a similar manner (Table 1). The typical yield was between 68 and 88% depending on the composition of the mixture (Table 2). An aliquot of the cooled reaction mixture was thoroughly analyzed by 1H-NMR, 13C-NMR and FTIR spectroscopy.| Condition | [M]0/[I]0 | [M]0/[cat]0 | Conversion (%) | Mn (SEC) (Da) | Mn (NMR) (g mol−1) | PDI | |
|---|---|---|---|---|---|---|---|
| D0 | 120 °C, 48 h | 333 | 333 | 68.1 | 5100 | 4550 | 1.39 |
| D1 | 120 °C, 48 h | 1000 | 1000 | 70.5 | 7240 | 7400 | 1.37 |
| D2 | 120 °C, 48 h | 2000 | 2000 | 69.5 | 8200 | 7150 | 1.41 |
| D3 | 120 °C, 48 h | 1000 | 2000 | 68.9 | 8730 | 6700 | 1.45 |
| D4 | 120 °C, 12 h | 794 | 1000 | 88.0 | 11700 |
8500 | 1.49 |
Fig. 1a shows typical 1H-NMR spectra of the poly-δ-valerolactone homopolymers. The signals obtained for the representative homopolymer D1 are: 1H-NMR (400 MHz, CDCl3, δ): 7.3 (s, 4H), 4.1 (m, 147H), 3.7 (t, 4H), 2.4 (m, 159H) and 1.7 (m, 330H). In 1H-NMR (Fig. 1a), the protons (e) adjacent to the acyl oxygen appearing at δ – 4.08–4.15 ppm, the protons (b) next to the carbonyl carbon appearing at δ – 2.3–2.4 ppm and the methylene protons (c and d) appearing at δ – 1.6–1.7 ppm are evidence of the formation of poly-δ-valerolactone. Only a small, low-intensity peak is noticed at δ – 3.9 ppm, indicating the presence of the terminal methylene (e) protons of the δ-valerolactone unit attached to the hydroxyl chain end. The participation of 1,6-hexanediol as the initiator is also apparent from the spectrum, which shows two terminal methylene resonances at δ – 3.6 ppm (1), but the other representative peaks (2,3) merge with the peaks for the protons (b–d) of poly-δ-valerolactone. The 1H-NMR spectra obtained for D0, D2, D3, D4 are similar to that of D1.
 | ||
| Fig. 1 (a) 1H-NMR spectra of δ-valerolactone homopolymers D0, D1, D2, D3 and D4. Inset shows a small peak at 3.9 ppm for D1. (b) 13C-NMR spectra of δ-valerolactone homopolymers D1, D2 and D4. Inset shows expanded carbonyl region for D1. | ||
The 13C-NMR spectra of the corresponding homopolymers are collated in Fig. 1b. The signals for the representative poly-δ-valerolactone homopolymer D1 are as follows: 13C-NMR (400 MHz, CDCl3, δ): 173.5, 171.6, 69.7, 68.1, 64.1, 33.9, 30.0, 28.2, 25.8, 22.5, 21.6 and 19.3. The signals assigned are: a carbonyl carbon (a) appearing at 173.0 ppm, a methylene carbon (e) adjacent to the acyl oxygen at 69.0 ppm, a methylene carbon (b) attached to a carbonyl group at 30.0 ppm and two other methylene carbons (c and d) appearing at 22.0 and 19.0 ppm. Thus the results of 13C-NMR spectroscopy are consistent with those of 1H-NMR spectroscopy, clearly indicating the success of the reaction. The incorporation of 1,6-hexanediol is also evident from the signals at 64.0 ppm showing the presence of the terminal methylene carbon (1) adjacent to an oxygen and the signals at 33.9 and 25.8 ppm attributed to the internal methylene carbons (2 and 3), respectively, of the 1,6-hexanediol unit. A quantitative up-field shift [e.g. from δ – 173.5 ppm (a) to δ – 171.69 ppm (a′) in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O region] has been observed for the segment of poly-δ-valerolactone, attached to the 1,6-hexanediol from the repeating segment. The 13C-NMR spectra of D0 (Fig. S1 ESI†), D2, D3 (Fig. S1 ESI†) and D4 also show all the representative peaks observed for D1.
O region] has been observed for the segment of poly-δ-valerolactone, attached to the 1,6-hexanediol from the repeating segment. The 13C-NMR spectra of D0 (Fig. S1 ESI†), D2, D3 (Fig. S1 ESI†) and D4 also show all the representative peaks observed for D1.
The characteristic frequencies observed for the δ-valerolactone monomer and a series of homopolymers in FTIR spectroscopy are shown in Fig. S2.† The broad region at 3530 cm−1 for OH stretching30 in the monomer increases in the spectra of the homopolymers, supporting the ring-opening of δ-valerolactone. The other characteristic vibrational frequencies, such as 1730 cm−1 for CO stretching, 2900 cm−1 for CH2 stretching, 1154–1172 cm−1 for the C–O–C bond and 1041 cm−1 for O–CH2 stretching validate the formation of poly-δ-valerolactone. These are in line with previously published data.31 The peak at 1185 cm−1 corresponding to C–O–C bond stretching in the monomer broadens with polymerization along with a small shift to 1180 cm−1, as clearly shown in Fig. S2 (ESI).† The band at 1106 cm−1, assigned to C–O–Sn bond formation,32 is present in all the δ-valerolactone homopolymers, but totally absent in the monomer. All these justify the success of the ring-opening polymerization.
After the precipitation of the polymer, the molar mass characteristics were measured by SEC (Fig. S3 ESI†) using polystyrene standards (Table 2). δ-Valerolactone with a variable block length was synthesized by altering the ratio of [M]0/[I]0 and [M]0/[cat]0 at a reaction temperature of 120 °C for 48 h. A monomodal mass distribution with PDI values ranging from 1.37 to 1.49 was observed for all the homopolymers. From the 1H-NMR spectra, the molecular weight of the homopolymer was calculated from the integral ratio of the methylene protons at δ – 4.1 ppm (e) of δ-valerolactone and the methylene protons of 1,6-hexanediol at δ – 3.6 ppm (1) (Fig. 1a). The value of Mn from the 1H-NMR spectra was in close proximity to that obtained from GPC (Table 2).
The effect of the catalyst concentration on the polymerization of δ-valerolactone was studied keeping the [M]0/[I]0 ratio constant (1000). The catalyst concentration [M]0/[cat]0 was varied from 1000 (for D1) to 2000 (for D3) and showed an increase in molecular weight and PDI as determined from SEC. The role of the initiator was evaluated when the catalyst concentration [M]0/[cat]0 was kept constant (1000). When the [M]0/[I]0 ratio decreased from 1000 (for D1) to 794 (for D4), a higher conversion and molecular weight were achieved in less time (12 h).
The optimization of the catalyst and initiator concentrations is crucial for polymerization. In this reaction, 1,6-hexanediol reacts with tin 2-ethyl hexanoate [Sn(Oct)2] to generate an alkoxide initiator, Sn(OR)2. It can also act as a chain transfer agent and can effectively control the molecular weight.33 If the concentration of Sn(Oct)2 increases, it leads to randomization in the copolymer sequencing. In this work, several polymerizations were carried out varying both [M]0/[I]0 and [M]0/[cat]0. All the parameters are tabulated in Table 2. The system with [M]0/[I]0 = 333 and [M]0/[cat]0 = 333 was found to be the least effective of all the systems in terms of conversion and molecular weight, whereas highest conversion at the shortest time was achieved with the [M]0/[I]0 = 794 and [M]0/[cat]0 = 1000 system.
A series of poly-D,L-lactide homopolymers (L1, L2, L3 and L4) was also synthesized and characterized (see ESI Fig. S4 and S5†). The 1H-NMR spectra of all the homopolymers show the representative peaks, confirming the ring-opening polymerization of poly-D,L-lactide. Fig. S4† shows typical 1H-NMR spectra of the poly-D,L-lactide homopolymers. The signals obtained for the representative homopolymer L1 are as follows: 1H-NMR (400 MHz, CDCl3, δ): 7.3 (s, 4H), 5.2 (m, 141H), 3.8 (t, 4H), 1.6 (m, 434H). In the 1H-NMR spectra (Fig. S4†), the methine protons (A) at δ = 5.2 ppm and the methyl protons (B) appearing at δ = 1.6 ppm are evidence of the formation of poly-D,L-lactide. Only a small, low-intensity peak is noticed at δ – 4.3 ppm, indicating the presence of the terminal methine (A) protons of the D,L-lactide unit attached to the hydroxyl chain end.6 The participation of 1,6-hexanediol as the initiator is also apparent from the spectrum, which shows two terminal methylene resonances at δ – 3.7 ppm (1); the other representative peaks (2 and 3) merge with the peaks for the protons (B) of poly-D,L-lactide.
The 13C-NMR spectra of the corresponding homopolymers are collated in Fig. S5.† The signals for the representative poly-D,L-lactide homopolymer L1 are as follows: 13C-NMR (400 MHz, CDCl3, δ): 169.6, 72.5, 69.4, 66.7, 20.5, 16.7, 15.8. The signals assigned are: a carbonyl carbon (C) appearing at 169.0 ppm, a methine carbon (A) at 69.0 ppm and a methyl carbon (B) at 16.0 ppm. Thus the results of the 13C-NMR spectra are consistent with those of 1H-NMR, clearly indicating the success of the reaction. The incorporation of 1,6-hexanediol is also evident from the signals at 66.0 ppm showing the presence of the terminal methylene carbon (1) adjacent to oxygen and 20.5 ppm, attributed to the internal methylene carbons (3) of the 1,6-hexanediol unit. As shown in Table S1 (ESI),† the GPC molecular weights of the various polymers lie in the range 11000–17000 Da. The NMR results corroborate the observations for GPC. These results are necessary for understanding the triblock copolymer made from D,L-lactide.
Synthesis and characterization of D,L-lactide–δ-valerolactone–D,L-lactide triblock copolymer
D,L-Lactide–δ-valerolactone–D,L-lactide copolymers were prepared by the ring-opening bulk polymerization technique using poly-δ-valerolactone as the macroinitiator and tin 2-ethyl hexanoate as the catalyst (Scheme 1). | ||
| Scheme 1 Sequential synthesis of D,L-lactide–δ-valerolactone–D,L-lactide triblock copolymer. | ||
The precipitated copolymers were analyzed by 1H-NMR, 13C-NMR and FTIR spectroscopy, and SEC. The 1H-NMR spectrum of a DL15050 copolymer is shown in Fig. 2a. The spectrum contains the signals of D,L-lactide as well as δ-valerolactone, as follows: 1H-NMR (400 MHz, CDCl3, δ): 7.3 (s, 4H), 5.2 (m, 240H), 4.1 (m, 535H), 2.3 (m, 588H), 1.7 (m, 1169H), 1.6 (m, 786H), 1.3 (s, 4H). The proton signals at 4.1, 2.3 and 1.6–1.7 ppm confirm the presence of the δ-valerolactone unit, as discussed earlier. The other two resonances at δ – 5.2 and 1.6 ppm account for the methine proton (A) and the methyl protons (B), respectively.
 | ||
| Fig. 2 (a) 1H-NMR spectrum of a representative copolymer, DL15050. (b) 1H-NMR spectrum of D1, showing signal around 3.9 ppm and 1H-NMR spectra for DL15050, DL16634, DL18020, showing signal around 4.3 ppm in the expanded region. (c) 13C-NMR spectrum of representative copolymer DL15050. (d) 13C-NMR spectra for copolymers, showing the expanded CO region (16–173 ppm). | ||
The terminal methylene protons (δ – 3.9 ppm) attached to the end hydroxyl group in poly-δ-valerolactone are replaced by the hydroxyl terminus methine proton of polylactide (PLA) (δ – 4.3 ppm) on chain extension,32 indicating quantitative chain initiation efficiency (Fig. 2a). In the course of the polymerization process, chain transfer can occur simultaneously34 where the active chain end of the PLA segment can undergo a trans-esterification reaction with the poly-δ-valerolactone unit, generating a δ-valerolactone chain end. This phenomenon will lead to the randomization of the chain segments, facilitating the formation of a random copolymer or a multiblock copolymer instead of the desired triblock. The absence of a δ-valerolactone chain end (δ – 3.9 ppm) (Fig. 2b) supported the fact that very limited trans-esterification had taken place.
Fig. 2c shows the 13C-NMR spectrum of a typical block D,L-lactide–δ-valerolactone–D,L-lactide copolymer, DL15050. The signals obtained for the corresponding copolymer are as follows: 13C-NMR (400 MHz, CDCl3, δ): 173.3, 169.6, 169.4, 169.1, 69.4, 69.2, 68.9, 68.3, 66.7, 65.0, 63.9, 33.7, 33.4, 33.1, 28.1, 27.9, 27.7, 21.4, 21.2, 20.5, 16.7, 16.6, 15.8 ppm. In the 13C-NMR spectrum the signals indicating the presence of the δ-valerolactone segment are as follows: 173.3 ppm for a carbonyl carbon (a), 68.0 ppm for a carbon (e) adjacent to the acyl oxygen, 28.1 ppm for a methylene carbon (b) attached to a carbonyl group; the other two internal methylene carbons (c and d) appeared at 21.4 and 20.5 ppm. The additional peaks for the LA repeating unit appeared at 169.5 ppm for CO (C), 69.0 ppm for the methine carbon (A) and 15.8 ppm for –CH3 (B), whereas the signals at 64.0 and 34.0 ppm establish the incorporation of 1,6-hexanediol in the main chain. This supports the formation of the copolymer. Multiple signals in the CO region (δ – 173.3, 169.6, 169.4, 169.1 ppm) confirm the block formation; 168–173 ppm represents the carbonyl signals for different monomer sequences.35 A sharp peak at 173.3 ppm and several low-intensity signals in the region of 169.0 ppm were observed for all the copolymers shown in Fig. 2d.
For the homopolymers poly-δ-valerolactone and poly-D,L-lactide, the CO resonances were observed at 173.7 and 169.1 ppm, respectively. However, in the copolymer, multiple peaks were observed instead of a single peak. A down-field shift of the additional carbonyl signals was observed for the lactyl unit due to the presence of the δ-oxyvalery unit appearing as triads LLV, VLL and VLV (Fig. 2d), whereas an additional up-field signal was seen for the carbonyl group of the δ-valerolactone unit corresponding to the triads LLV and LVL.36 To establish the formation of the triblock, we synthesized a copolymer of δ-valerolactone and D,L-lactide by simultaneous addition (DVL-S-DLLA). A comparison of the 13C-NMR spectra in the carbonyl region between DVL-S-DLLA and all the copolymers synthesized by step addition showed (Fig. 2d) that there were additional peaks in the case of DVL-S-DLLA. These additional peaks, generated due to random cross-propagation reactions between the D,L-lactide and δ-valerolactone active chain ends, were completely absent in the synthesized block copolymers. Thus the analysis of the CO signals of the copolymer verifies the triblock sequence of the δ-oxyvalery and lactyl units. As the δ-valerolactone monomer is absent in the second step of polymerization, trans-esterification is the only route that can lead to randomization of the chain sequence. The existence of the CO signal at 170 ppm for the VLV triad gives evidence for trans-esterification37 and the extent of trans-esterification is represented quantitatively by the relative intensity of the VLV triad. No trans-esterification occurs in the case of DL15050, DL16634 and DL18020. For other triblock copolymers, the extent of trans-esterification calculated is as follows: for DL25050, 7.2%; for DL26634, 13.3%; for DL28020, 14.6%; for D2L1, 9.4%; and for D2L3, 10.1%. Therefore limited trans-esterification was observed for all the synthesized block copolymers, as confirmed by the 13C-NMR spectra.
FTIR analysis of copolymer
The FTIR spectra of a series of triblock copolymers (DL15050, DL16634 and DL18020) were compared with the corresponding homopolymer (D1) and monomer (Fig. S6, ESI†). The synthesized copolymer contained all the representative peaks for δ-valerolactone and D,L-lactide. The broad region at 3560 cm−1 for OH stretching increased on copolymerization, suggesting the success of the reaction. νCH3 (2965 cm−1), the representative frequency of D,L-lactide, overlapped with the νCH2 of δ-valerolactone. The peak areas corresponding to the signals at 1326 cm−1 for C–H bending and at 1730 cm−1 for CO stretching showed an increase on polymerization. The peak at 1106 cm−1, attributed to C–O–Sn bond formation, was absent in the copolymers DL15050, DL16634 and DL18020, indicating the replacement of Sn. The band at 1176 cm−1 for C–O–C stretching in D1 shifted to 1187 cm−1 for DL15050, but remained the same for the others. The other relevant vibrational frequencies, such as νCO, νCOO, were similar to those for δ-valerolactone.
GPC analysis
Comparison between the SEC chromatograms of the poly-δ-valerolactone prepolymer and the D,L-lactide–δ-valerolactone–D,L-lactide triblock copolymers is shown in Fig. S7 (ESI).† The shift in the chromatograms of the copolymers towards a shorter retention time confirmed the chain extension reaction in the presence of the D,L-lactide monomer. In this work, poly-δ-valerolactone was synthesized first, then it acted as a bifunctional hydroxyl-capped macroinitiator for the ring-opening polymerization of D,L-lactide, leading to the formation of the triblock D,L-lactide–δ-valerolactone–D,L-lactide. Table 3 shows the feed composition, copolymer composition, Mn and PDI for a series of the copolymers. The poly-δ-valerolactone and D,L-lactide feed ratio was varied to alter the block length of D,L-lactide. DL15050, DL16634 and DL18020 were synthesized using D1 as the macroinitiator. The Mn value for these copolymers ranged between 10433 and 11056 Da. For the other series DL25050, DL26634 and DL28020 using D2 as the macroinitiator, there was a sharp increase in the Mn as the wt% of D,L-lactide was increased in the copolymer composition. For D2L1, D2L2 and D2L3, the macroinitiator was D2, but the [M]0/[cat]0 ratio was varied from 5000 to 794 in the second step of the polymerization, i.e. the addition of D,L-lactide, while keeping the monomer ratio at 66:34. As the [M]0/[cat]0 ratio decreased, Mn increased. Among the homopolymers, D1 and D2 were used as macroinitiators for further reaction with D,L-lactide with a feed ratio of 50:50. The molecular weight (from SEC) of DL15050 was little higher than that of DL25050. DL15050 contains more of the δ-valerolactone segment (61%) than DL25050 (59%).
| wt% of lactide in monomer (theoretical) | wt% of lactide in polymer (experimental) | wt% of δ-valerolactone in monomer (theoretical) | wt% of δ-valerolactone in polymer (experimental) | Molecular weight (g mol−1) determined from SEC | PDI | |
|---|---|---|---|---|---|---|
| D0 | — | — | — | — | 5100 | 1.39 |
| D1 | — | — | — | — | 7240 | 1.23 |
| DL15050 | 50 | 39 | 50 | 61 | 10400 |
1.32 |
| DL16634 | 34 | 22 | 66 | 78 | 11000 |
1.49 |
| DL18020 | 20 | 10 | 80 | 90 | 10600 |
1.45 |
| D2 | 8200 | 1.85 | ||||
| DL25050 | 50 | 41 | 50 | 59 | 9400 | 1.23 |
| DL26634 | 34 | 32 | 66 | 68 | 8300 | 1.40 |
| DL28020 | 20 | 12 | 80 | 88 | 5500 | 1.37 |
| D2L1 | 34 | 20 | 66 | 80 | 7680 | 1.39 |
| D2L2 | 34 | 32 | 66 | 68 | 9400 | 1.23 |
| D2L3 | 34 | 23 | 66 | 77 | 10100 |
1.33 |
Wide-angle X-ray diffraction
Fig. 3a and b show the X-ray diffractograms of the homopolymers and copolymers with variable compositions of δ-valerolactone and D,L-lactide. All the WAXD profiles show well-resolved diffraction peaks in the 2θ range 0–40°. The X-ray diffractogram for poly-D,L-lactide shows a broad hump, indicating the amorphous nature of the polymer, whereas one intense major peak at 21.7° and a smaller peak at 24.4° are observed for poly-δ-valerolactone. The crystallographic analysis clarified that poly-δ-valerolactone acquired an orthorhombic unit cell structure.38 The homopolymers D0, D1 and D2 have a crystallinity of 33.3, 28.5 and 29.8%, respectively. In these copolymers, similar diffraction patterns, indicating the contribution of crystalline poly-δ-valerolactone, are observed. However, the presence of D,L-lactide was reflected in a reduction of the crystallinity on increasing the D,L-lactide concentration in the copolymer. The incorporation of D,L-lactide reduced the crystallization ability of poly-δ-valerolactone39 (Fig. 3c). There was a sharp increase in the Mn as the D,L-lactide composition increased in the copolymer (Table 3), but the crystallinity showed a reverse trend. The decrease in crystallinity with increasing D,L-lactide block length was due to the amorphous nature of the D,L-lactide (Fig. 3c). | ||
| Fig. 3 (a) WAXD patterns for homopolymers D1, D2 and D3. (b) Comparison of WAXD patterns between homopolymer poly-δ-valerolactone (PVL) and poly-D,L-lactide (PLA) and copolymers DL15050, DL16634 and DL18020. (c) Variation in percentage crystallinity with increasing concentrations of δ-valerolactone for copolymers based on D0, D1 and D2. | ||
The crystallite size was calculated using the Scherrer method and the lattice strain was determined using the Williamson–Hall isotropic strain model (W–H-ISM).40
The Scherrer equation gives the mean crystallite size as follows:
|
Lc = kλ/βcosθ
| (1) |
The W–H-ISM is framed considering two factors, crystallite size and lattice strain, assuming that the strain in the crystal lattice is uniform:
|
β cosθ = kλ/Lc + 4εsinθ
| (2) |
cosθ on the y-axis versus 4sinθ on the x-axis gives the magnitude of the lattice strain (ε) as the slope; the crystallite size, Lc, is evaluated from the intercept. All the values of lattice strain, crystallite size and percentage crystallinity for the polymers are given in Table 4.
| Sample | Crystallite size | Lattice strain (W–H method) (×10−3) | Crystallinity (%) | |
|---|---|---|---|---|
| Scherrer method | ||||
| 2θ (°) | Lc (nm) | |||
| D1 | 21.8 | 18.64 | 3.41 | 28.5 |
| DL18020 | 21.7 | 16.41 | 4.69 | 24.0 |
| DL16634 | 21.8 | 16.50 | 9.89 | 19.5 |
| DL15050 | 21.8 | 16.78 | 13.22 | 13.7 |
| D2 | 21.8 | 17.66 | 3.82 | 29.8 |
| DL28020 | 21.7 | 16.44 | 6.17 | 24.0 |
| DL26634 | 21.8 | 16.47 | 6.36 | 17.0 |
| DL25050 | 21.7 | 18.09 | 6.54 | 15.0 |
| D0 | 21.8 | 25.93 | 12.97 | 33.3 |
| DL7030 | 21.8 | 16.89 | 12.37 | 19.4 |
| DL6040 | 21.7 | 19.68 | 19.68 | 21.3 |
| DL4060 | 21.8 | 34.14 | 13.04 | 12.6 |
The crystallite size in general increases as the block length of the D,L-lactide segment increases and the lattice strain follows the same trend.
Thermal analysis
The DSC thermograms of some representative samples of the D,L-lactide–δ-valerolactone–D,L-lactide triblock copolymer are shown in Fig. 4 and the glass transition temperatures of the copolymers are given in Table 5. The data showed two glass transition temperatures, indicating the presence of two blocks. The glass transition temperature of poly-δ-valerolactone appeared around −62.0 °C and for poly-D,L-lactide the Tg was around 40.5 °C. All the copolymers showed two glass transition temperatures which shifted depending on the block length of the δ-valerolactone and D,L-lactide segments. The Tg of the δ-valerolactone segment shifts towards higher values (in the range −47.0 to −33.0 °C), whereas the Tg of the D,L-lactide segment shifts towards lower values (in the range −33.0 to 4.19 °C). The higher block length of poly-D,L-lactide shifts the Tg of δ-valerolactone towards higher values, clearly indicating that the Tg of the block copolymer depends on the segmental length of the poly-D,L-lactide. All the copolymers melt in the range 51–66 °C.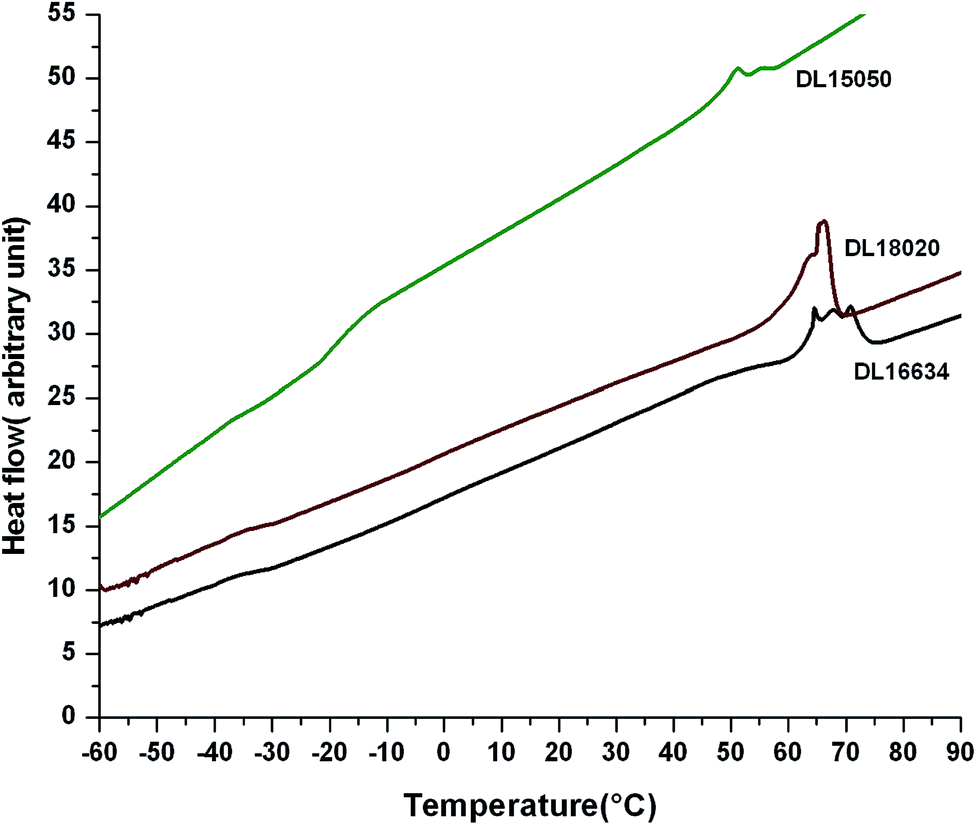 | ||
| Fig. 4 DSC analysis for DL16634, DL18020 and DL15050. | ||
| Sample | Tg (δ-valerolactone) (°C) | Tg (D,L-lactide) (°C) |
|---|---|---|
| D1 | −62.0 | — |
| L1 | — | 40.5 |
| DL15050 | −32.8 | −12.4 |
| DL16634 | −41.2 | −32.7 |
| DL18020 | −46.8 | −32.4 |
| DL25050 | −37.4 | −22.2 |
| DL26634 | −36.3 | −3.1 |
| DL28020 | −52.7 | −29.3 |
| DL4060 | −35.6 | 20.8 |
| DL6040 | −66.9 | 4.2 |
Multimodal melting endotherms were observed for the copolymers, suggesting a broad distribution of the crystallite size. This occurs due to microphase separation in the melt state as a result of chemical incompatibility between the blocks, followed by crystallization. When the microstructure of the copolymer is solely crafted by the crystallization of one block, the crystallizable component can be subdivided, forming microdomains which appear to be higher in number than the number of active heterogeneities present. The multimodal melting endotherm is mainly evident for the low molecular weight copolymer.41
Morphological analysis
The TEM micrographs of DL15050, DL16634 and DL18020 in toluene and DL15050 in chloroform are shown in Fig. 5. All the samples show a uniform lamellar morphology with stripes of alternating contrast. From the WAXD study it is evident that the block copolymer contains amorphous poly-D,L-lactide domains and crystalline poly-δ-valerolactone domains. During staining, the amorphous regions of poly-D,L-lactide were stained preferentially and appeared dark; the poly-δ-valerolactone segment appeared bright under TEM. The domain size ranges between 3 and 6 nm. The TEM images obtained are similar to those observed previously for a poly-L-lactide–polymenthide–poly-L-lactide triblock copolymer.41 However, the domain size is much lower for poly-L-lactide–poly-δ-valerolactone–poly-D,L-lactide copolymer compared with the poly-L-lactide–polymenthide–poly-L-lactide triblock copolymer.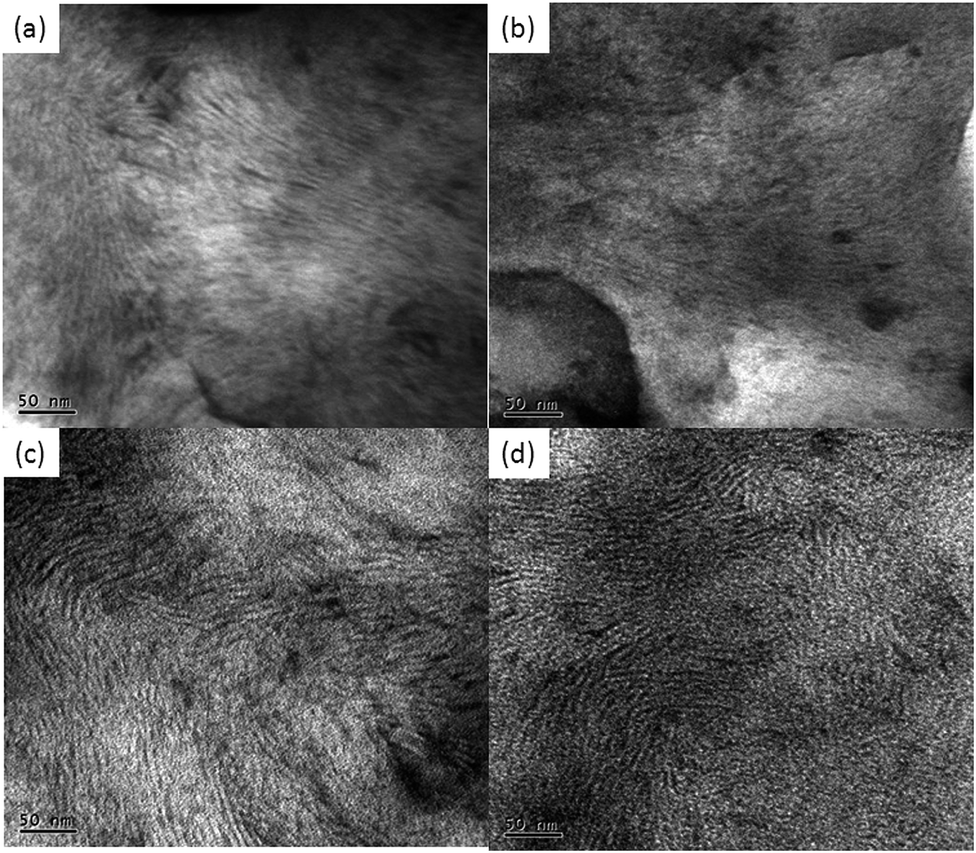 | ||
| Fig. 5 TEM micrographs of triblock copolymer: (a) DL15050 in toluene; (b) DL16634 in toluene; (c) DL18020 in toluene; and (d) DL15050 in chloroform. | ||
Characterization of microspheres loaded with salicylic acid
The FESEM micrographs of microspheres loaded with salicylic acid, taken before and after release, are shown in Fig. 6. The microspheres adopted a regular spherical morphology with an average diameter of 17.2 μm. Before release, the surface of the microspheres shows a smooth morphological pattern (Fig. 6b). In the buffer medium (PBS, pH 7.4), the salicylic acid is released, resulting a porous appearance to the surface of the microsphere. The small pores (Fig. 6d) have dimensions in the range 1–4 μm, supporting the suggestion that the release of salicylic acid is favored by the degradation of the copolymer on the surface of the microsphere. | ||
| Fig. 6 SEM micrographs: (a) microsphere loaded with salicylic acid for DL16634; (b) surface of microsphere loaded with salicylic acid before release; (c) surface of microsphere loaded with salicylic acid after release; and (d) pores generated on microsphere surface after release. | ||
The FTIR spectra (Fig. 7) give evidence of the encapsulation of salicylic acid, showing a peak at 1617 cm−1 for aromatic CC stretching without a distinct shift, which indicates that no chemical interaction has taken place between the copolymer and salicylic acid. Pristine salicylic acid showed a maximum degradation temperature around 198 °C (Fig. S8 inset, ESI†) and the heat flow curve, as well as the derivative thermogram of microspheres loaded with salicylic acid, showed a hump around 202 °C, indicating the degradation of salicylic acid. Thermogravimetric analysis of the degradation curve confirmed 20% loading of salicylic acid within the microsphere at 202 °C (Fig. S8, ESI†). The release profile for microspheres loaded with salicylic acid in PBS was studied for two samples (DL16634 and DL18020) and the results are summarized in Fig. 8. For both samples, a burst release was initially observed, which increased with time and became steady after 24 h. For DL16634, 10.3% was released after 2 h, which gave 12.2% after 24 h. The release rate of the drug from DL28020 loaded with salicylic acid was slower than that of DL16634 i.e. 8.8% after 2 h and only 10.1% after 24 h. An initial burst release probably occurred because the drug was attached to the surface of the microsphere. Later, the release is governed by diffusion,42 which is suppressed by the hydrophobic nature of the copolymer inhibiting the diffusion of water into the core and the diffusion of salicylic acid into the medium.
 | ||
| Fig. 7 FTIR spectra of microspheres, salicylic acid-loaded microspheres, and salicylic acid. | ||
 | ||
| Fig. 8 Release profile of salicylic acid from microsphere. | ||
Conclusions
A novel D,L-lactide–δ-valerolactone–D,L-lactide copolymer was successfully synthesized and characterized by various techniques. The sequential synthesis procedure adopted for the D,L-lactide–δ-valerolactone–D,L-lactide showed the formation of a hydroxyl-end functionalized poly-δ-valerolactone by the presence of small peak at 3.9 ppm, detected by 1H-NMR, which disappeared with the addition of the D,L-lactide moiety in the second step of the polymerization. A peak at 4.3 ppm, corresponding to the D,L-lactide chain end, appeared in the 1H-NMR spectra. The success of the reaction was also ascertained by the broadening of peaks in the OH-stretching region at 3560 cm−1 and the carbonyl region at 1730 cm−1. The reaction conditions for the synthesis of poly-δ-valerolactone were optimized in terms of the initiator and catalyst concentrations and the ring-opening polymerization of δ-valerolactone at 120 °C for 12 h; [M]0/[I]0 = 794 and [M]0/[cat]0 = 1000 were found to be the most effective. The block sequencing determined from the carbonyl region of the 13C-NMR spectra showed that no trans-esterification was observed for DL15050, DL16634 and DL8020. Thorough analysis of the 13C-NMR and GPC results showed that the molecular weight varied as a function of the block length, initiator to catalyst ratio and the extent of trans-esterification. The molecular weight increased as the ratio of the D,L-lactide segment increased, but, as a result of its amorphous nature, it reduced the crystallinity, as evident from WAXD analysis. DSC showed two glass transition temperatures, a characteristic feature of a block copolymer. However, the glass transition temperatures shifted towards lower or higher values when the block length of the copolymers was varied. The copolymer adopted a lamellar morphology with alternating stripes, showing the microphase separation, where the more crystalline poly-δ-valerolactone moiety appeared as a bright region and the comparatively darker domains were assigned to the amorphous poly-D,L-lactide segment. It was confirmed from the thermogravimetric analysis and FTIR spectra that the triblock microsphere could successfully encapsulate salicylic acid. In the presence of a buffer medium (PBS pH 7.4), the salicylic acid was released from the microsphere, forming small holes on its surface, and the release profile clearly showed its potential as a drug-delivery system.References
- E. Llorens, M. M. Perez-Madrigal, E. Armelin, L. J. del Valle, J. Puiggali and C. Aleman, RSC Adv., 2014, 4, 15245–15255 RSC
.
- Y.-S. He, J.-B. Zeng, G.-C. Liu, Q.-T. Li and Y.-Z. Wang, RSC Adv., 2014, 4, 12857–12866 RSC
- A. Ganguly, A. K. Bhowmick and Y. Li, Macromolecules, 2008, 41, 6246–6253 CrossRef CAS
- S. Ghosh, A. K. Bhowmick, N. Roychowdhury and G. Holden, J. Appl. Polym. Sci., 2000, 77, 1621–1628 CrossRef CAS
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed
- T. Li, T. Ci, L. Chen, L. Yu and J. Ding, Polym. Chem., 2014, 5, 979–991 RSC
- A. Parthiban, A. Likhitsup, F. M. Coo and C. L. L. Chai, Polym. Chem., 2010, 1, 333–338 RSC
- S. I. Jeong, B.-S. Kim, Y. M. Lee, K. J. Ihn, S. H. Kim and Y. H. Kim, Biomacromolecules, 2004, 5, 1303–1309 CrossRef CAS PubMed
- R. R. Gowda and D. Chakraborty, J. Mol. Catal. A: Chem., 2010, 333, 167–172 CrossRef CAS PubMed
- Y. Nakayama, K. Sasaki, N. Watanabe, Z. Cai and T. Shiono, Polymer, 2009, 50, 4788–4793 CrossRef CAS PubMed
- N. Kumar, M. N. V. Ravikumar and A. J. Domb, Adv. Drug Delivery Rev., 2001, 53, 23–44 CrossRef CAS
- X. Lou, C. Detrembleur and R. Jérôme, Macromolecules, 2002, 35, 1190–1195 CrossRef CAS
- J. E. Báez, M. Martínez-Rosales and A. Martínez-Richa, Polymer, 2003, 44, 6767–6772 CrossRef PubMed
- A. Nakayama, N. Kawasaki, I. Arvanitoyannis, J. Iyoda and N. Yamamoto, Polymer, 1995, 36, 1295–1301 CrossRef CAS
- K. Stridsberg, M. Ryner and A.-C. Albertsson, Adv. Polym. Sci., 2002, 157, 41–65 CrossRef CAS
- H. Lee, F. Zeng, M. Dunne and C. Allen, Biomacromolecules, 2005, 6, 3119–3128 CrossRef CAS PubMed
- F. Faÿ, E. Renard, V. Langlois, I. Linossier and K. Vallée-Rehel, Eur. Polym. J., 2007, 43, 4800–4813 CrossRef PubMed
- F. Hironobu, Y. Masaru, A. Masaharu, K. Minoru, M. Tooru, Y. Hisako, I. Kyoichi, Y. Hidetoshi, K. Umeko and S. Keiji, J. Controlled Release, 1989, 10, 293–303 CrossRef
- S. K. Bhattacharyya and D. K. Nandi, Ind. Eng. Chem., 1959, 51, 143–146 CrossRef CAS
- Y.-C. Chang and I. M. Chu, Eur. Polym. J., 2008, 44, 3922–3930 CrossRef CAS PubMed
- Z. L. Tyrrell, Y. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128–1143 CrossRef CAS PubMed
- R. Yang, F. Meng, S. Ma, F. Huang, H. Liu and Z. Zhong, Biomacromolecules, 2011, 12, 3047–3055 CrossRef CAS PubMed
- H. M. Wong, J. J. Wang and C.-H. Wang, Ind. Eng. Chem. Res., 2001, 40, 933 CrossRef CAS
- H. Okada and H. Toguchi, Crit. Rev. Ther. Drug Carrier Syst., 1995, 12, 1–99 CrossRef CAS
- G. Li, Q. Cai, J. Bei and S. Wang, Polym. Adv. Technol., 2003, 14, 239–244 CrossRef CAS
- K. L. Nair, S. Jagadeeshan, S. A. Nair and G. S. Kumar, J. Nanobiotechnol., 2011, 9, 42–56 CrossRef PubMed
- W.-J. Lin, C.-L. Wang and L.-W. Juang, J. Appl. Polym. Sci., 2006, 100, 1836–1841 CrossRef CAS
- E. M. Frick, A. S. Zalusky and M. A. Hillmyer, Biomacromolecules, 2003, 4, 216–223 CrossRef CAS PubMed
- G. Grancharov, O. Coulembier, M. Surin, R. Lazzaroni and P. Dubois, Macromolecules, 2010, 43, 8957–8964 CrossRef CAS
- W. Dai, J. Zhu, A. Shangguan and M. Lang, Eur. Polym. J., 2009, 45, 1659–1667 CrossRef CAS PubMed
- W. Saiyasombat, R. Molloy, T. M. Nicholson, A. F. Johnson, I. M. Ward and S. Poshyachinda, Polymer, 1998, 39, 5581–5585 CrossRef CAS
- C.-S. Wu, J. Appl. Polym. Sci., 2004, 92, 1749–1757 CrossRef CAS
- Y. Baimark and R. Molloy, ScienceAsia, 2004, 30, 327–334 CrossRef CAS
- M. T. Martello and M. A. Hillmyer, Macromolecules, 2011, 44, 8537–8545 CrossRef CAS
- D. W. Grijpma, G. J. Zondervan and A. J. Pennings, Polym. Bull., 1991, 25, 327–333 CrossRef CAS
- H. Fukuzaki, M. Yoshida, M. Asano, Y. Aiba and I. Kaetsu, Eur. Polym. J., 1988, 24, 1029–1036 CrossRef CAS
- D. W. Grijpma and A. J. Pennings, Polym. Bull., 1991, 25, 335–341 CrossRef CAS
- Y. Furuhashi, P. Sikorski, E. Atkins, T. Iwata and Y. Doi, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2622–2634 CrossRef CAS
- Z. Zhao, L. Yang, Y. Hu, Y. He, J. Wei and S. Li, Polym. Degrad. Stab., 2007, 92, 1769–1777 CrossRef CAS PubMed
- N. Roy and A. K. Bhowmick, J. Phys. Chem. C, 2012, 116, 8763–8772 CAS
- C. L. Wanamaker, M. J. Bluemle, L. M. Pitet, L. E. O'Leary, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2904–2911 CrossRef CAS PubMed
- Y. Hu, X. Jiang, Y. Ding, L. Zhang, C. Yang, J. Zhang, J. Chen and Y. Yang, Biomaterials, 2003, 24, 2395–2404 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of poly-D,L-lactide, FTIR spectra, GPC chromatograms, NMR spectra, TGA thermogram, table. See DOI: 10.1039/c4ra02745k |
| This journal is © The Royal Society of Chemistry 2014 |