DOI:
10.1039/C4RA02736A
(Paper)
RSC Adv., 2014,
4, 24222-24231
Controlled delivery of dexamethasone to the intestine from poly(vinyl alcohol)–poly(acrylic acid) microspheres containing drug-cyclodextrin complexes: influence of method of preparation of inclusion complex
Received
28th March 2014
, Accepted 22nd May 2014
First published on 23rd May 2014
Abstract
pH-sensitive interpenetrating polymeric network hydrogel microspheres comprised of poly(vinyl alcohol) and poly(acrylic acid) were synthesized for delivery of dexamethasone (DX) to the intestine. DX is highly active in the treatment of virtually every type of B-cell malignancy; however significant side-effects are invariably associated with it thereby warranting a delivery system that can deliver DX at a controlled rate at the therapeutic level at a specific site. To regulate the release rate of DX, a preformed solid inclusion complex of DX with β-cyclodextrin was added into the hydrogel. In order to find out the influence of the method of preparation of the inclusion complex on the drug delivery process, inclusion complexes were prepared by co-precipitation and freeze-drying methods. Microspheres containing free drug, the physical mixture and the inclusion complexes were synthesized. The microspheres exhibited negligible drug release in the simulated gastric fluid but significant release in the intestinal fluid. The cytotoxicity assay ensured that the microspheres were biocompatible. Thus the synthesized microspheres could be effectively employed for the oral delivery of dexamethasone and their pH sensitivity could be exploited for the intestinal delivery. The microspheres containing the freeze-dried inclusion complexes were found to be the best of the lot for achieving a controlled release of DX. Therefore, it can be proposed that the adverse side-effects of DX could be minimized by using these microspheres as delivery vehicles.
1. Introduction
The design of oral drug delivery systems targeting the lower part of the gastrointestinal tract has gained momentum for the delivery of an array of therapeutic agents.1,2 While the stomach is hostile for drugs due to its low pH; the lower gut such as the intestine and colon, as sites of drug delivery, offer numerous therapeutic advantages because of their near neutral pH and longer transit time.3,4 Intestine including the colon is susceptible to certain diseases such as Crohn's disease, irritable bowel syndrome, ulcerative colitis, colon cancer, polyps etc. which require localized drug delivery.5 Various polymeric based devices have been proposed for such site-specific delivery of drug for efficient therapy.6–10 pH-sensitive smart hydrogels have garnered special interest in this context.11,12 Hydrogels composed of natural polysaccharides such as chitosan or synthetic polymers like poly(acrylic acid) and poly(methacrylic acid) have been employed in the fabrication of pH-sensitive formulations.13–15 The pH-sensitivity of these hydrogels is due to the presence of weakly acidic/basic functional groups in the polymer whose water-uptake properties depends on the pH of the external medium.14 Poly(acrylic acid) (PAA) has been widely employed because of its strong muco-adhesive and biocompatible properties.16 However, the application of pure PAA is limited because of the fast release of the drug due to its extensive swelling in water. Moreover, PAA tend to dissolve at high pH solution. The most commonly used strategy to alleviate these problems is to prepare three-dimensional polymeric network by physical or chemical crosslinking or incorporation of PAA in an interpenetrating polymeric network (IPN).17 Poly(vinyl alcohol) (PVA) hydrogels are widely utilized in biomedical and pharmaceutical fields because of their biocompatibility, biodegradability and non-toxic nature.18–20 Moreover, PVA is one of the most preferred component for synthesis of IPNs because of its long term temperature and pH stability. Hydrogels based on PVA and PAA have emerged as promising materials in biomedical applications due to their highly tunable chemical and physical properties.21–24
Dexamethasone (DX), a potent synthetic glucocorticoid, is frequently used to treat a wide spectrum of autoimmune and inflammatory diseases. This is one of the most preferred corticosteroid to treat inflammatory bowel disease, Crohn's disease and ulcerative colitis, which need its topical administration and often administered as enema. But in this method the drug gains access only to the rectum and the descending colon. Therefore, a number of approaches are being developed to deliver steroids to the intestine and colon via the oral route.25,26 Moreover significant side-effects are invariably associated with DX.27,28 In addition, it is a lipophilic drug (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 1.9) with extremely low aqueous solubility (0.16 mg mL−1) as a result the bioavailability of DX is lowered and its efficiency is hindered.29 Thus the formulation for this drug particularly is more challenging and warrants a delivery system that can deliver DX at a controlled rate to the specific site. Cyclodextrins have the potential to control the rate of drug release from polymeric matrices when grafted or covalently bound to the polymer backbone.30 They also have the potential to reduce the release rate of drug from hydrogels even without any covalent linking to the matrices.6,18,31 In our earlier works, it has been demonstrated that the addition of preformed drug-cyclodextrin inclusion complexes directly to the hydrogel matrix significantly controls the rate of drug release.6,18
P = 1.9) with extremely low aqueous solubility (0.16 mg mL−1) as a result the bioavailability of DX is lowered and its efficiency is hindered.29 Thus the formulation for this drug particularly is more challenging and warrants a delivery system that can deliver DX at a controlled rate to the specific site. Cyclodextrins have the potential to control the rate of drug release from polymeric matrices when grafted or covalently bound to the polymer backbone.30 They also have the potential to reduce the release rate of drug from hydrogels even without any covalent linking to the matrices.6,18,31 In our earlier works, it has been demonstrated that the addition of preformed drug-cyclodextrin inclusion complexes directly to the hydrogel matrix significantly controls the rate of drug release.6,18
With this rational we aimed at designing a delivery system for controlled delivery of DX to the intestine. To achieve specific delivery to the intestine, pH-sensitive sequential IPN hydrogel microspheres comprising of PVA and PAA were synthesized using glutaraldehyde as cross linker. The solid inclusion complexes of DX with β-cyclodextrin (CD) were added to the hydrogel matrix in order to get a controlled release. There are various methods of forming inclusion complexes and the physico-chemical properties of the complexes are strongly dependent on the method of preparation.32 Therefore, it would be interesting to find out whether the method of preparation of inclusion complex will influence the drug release process from hydrogels. For this reason the solid inclusion complexes of DX with CD were prepared by two commonly used methods; co-precipitation and freeze-drying. Microspheres containing the free drug, the drug-CD physical mixture, the co-precipitated inclusion complex and the freeze-dried inclusion complex were synthesized, characterized and their drug delivery potential was explored. The preliminary kinetics of drug release from these microspheres was investigated to have an approximate idea about the mechanism of drug transport. Cytotoxicity assay has been performed to inspect their biocompatibility.
2. Materials and methods
2.1 Materials
PVA (Mw = 89000–98000; 99.0% hydrolyzed), CD and DX were procured from Sigma-Aldrich, India. Ceric ammonium nitrate (CAN), light liquid paraffin oil and hydrochloric acid were obtained from S. D. Fine Chemicals, Mumbai, India. Acrylic acid (AAc) was purchased from SRL, India. Tween-80 and hexane were received from Merck, India. Glutaraldehyde (GA) (25% w/v) was supplied by Spectrochem Pvt. Ltd, Mumbai, India. Triply distilled water was utilized throughout.
2.2. Preparation of solid inclusion complexes
The preparation of solid inclusion complexes of DX and CD was performed by two different methods. The molar ratio of DX to CD was maintained at 1:1 in all cases.
Co-precipitation method33. Briefly, CD (0.2 g) was dissolved in distilled water at 50 °C for 1 h. DX solution (0.07 g) in ethanol was slowly added to the CD solution and the mixture was continuously stirred for 5 h. The final solution was refrigerated for 12 h at 4 °C. The co-precipitated DX–CD inclusion complex (CP) was recovered by filtration and washed with ethanol to remove the free DX. The residue was vacuum-dried and stored in air tight bottles.
Freeze drying method34. The freeze dried inclusion product (FD) was prepared by adding stoichiometric amounts of drug (0.07 g) to CD (0.2 g) solution and stirring for 5 h. The resulting solution was frozen at −20 °C and then lyophilized for 48 h (Scanvac Coolsafe Lyophilizer).
Physical mixture. A physical mixture (PM) of DX (0.07 g) and CD (0.2 g) was obtained in 1:1 molar ratio by homogeneous blending in a mortar for 15 min.
2.3. Synthesis of microspheres
Synthesis of PVA–PAA microspheres was carried out according to reported method.35 CAN have been used as an initiator for the polymerisation of AAc.36,37 10 wt% PVA solution was prepared by dissolving in distilled water at 80 °C for 4 h. Requisite amount of AAc monomer (10 wt%) was taken in distilled water and added drop-wise to the PVA solution with continuous stirring maintained at 50 °C. Then 0.2 g CAN was dissolved in 10 mL water and added to the above solution and vigorously stirred. The solution was cooled down to room temperature followed by the addition of free drug or the inclusion complex and stirred for 30 min to get a homogeneous mixture. The entire solution was emulsified to form water-in-oil (w/o) emulsion in 100 mL of light liquid paraffin oil containing 2% (w/v) Tween-80, 1 mL of 0.1 M HCl and 1 mL of 0.1 M GA and stirred for 5 h. The microspheres formed were centrifuged and washed repeatedly with hexane and water to remove the excess of reactants. The microspheres were dried under vacuum and stored in desiccator for further use. Blank microspheres without containing any drug were also synthesized for comparison and are labelled as MS1. The DX-loaded microspheres are designated MS2 and the PM loaded microspheres are labelled MS3. The CP and FD loaded microspheres are marked MS4 and MS5, respectively.
2.4. Instruments for characterization
FTIR. The FTIR spectra were recorded on a Perkin Elmer RX I spectrophotometer from 4000 to 400 cm−1 using KBr as reference at room temperature.
XRD. The XRD profiles were collected on a PANalytical X-ray diffractometer using nickel-filtered Cu Kα radiation and scanned from 5° to 40° at room temperature at a scan rate of 3° min−1.
DSC. DSC was performed by Mettler Toledo DSC822 instrument on 10 mg of samples under N2 atmosphere (purging rate: 40 mL min−1) from 50–300 °C at a heating rate of 10 °C min−1.
SEM. The SEM micrographs of the samples were observed on a JEOL SEM, JSM 6480LV model. The samples were sputtered coated with gold, mounted on metal stubs and then observed under SEM.
1H NMR. 1H NMR experiments were carried out in D2O obtained from Sigma-Aldrich, India. The spectra were acquired on a Bruker 400 MHz NMR at 298 K.
2.5. Drug loading efficiency
Estimation of drug concentration was done as per the reported method.38 Microspheres of known weight (10 mg) were finely ground using a mortar, extracted with 50 mL of distilled water and sonicated for 30 min (Electrosonic Industries, India). The solution was centrifuged (Remi Research Centrifuge, India) to remove the polymeric debris and the clear supernatant was analyzed using UV-vis spectrophotometer (Shimadzu, UV-1800) at λmax = 242 nm. The percent drug loading were calculated as:| |
 | (1) |
The values reported are the mean of the three independent measurements.
2.6. Swelling properties of microspheres
The swelling of MS1 was studied in solutions of pH 1.2 (dilute HCl) and pH 7.4 (phosphate buffer) at the physiological temperature of 37 °C. The microspheres were allowed to swell and weighed at regular interval of time till a constant weight was achieved. The percentage equilibrium swelling was calculated as:| |
 | (2) |
where Weq and Wd are the swollen and dry weights of the microspheres respectively. The values reported are the mean of the three independent measurements.
2.7. In vitro drug release studies
In vitro drug release studies from the microspheres were carried out at pH 1.2 (dilute HCl) and 7.4 (phosphate buffer). The drug release studies were also carried out in simulated gastric (SGF) and simulated intestinal fluids (SIF) according to standard methods reported in US Pharmacopeia. Weighed quantities of the drug loaded microspheres were placed in 50 mL of the releasing medium at a temperature of 37 °C. Then 3 mL aliquots were withdrawn from the samples at particular time intervals. The displaced medium was replenished with 3 mL of fresh solution so as to maintain sink conditions. The amount of DX released was estimated spectrophotometrically at a fixed λmax value of 242 nm. The release data are expressed as the mean value of three independent experiments and the standard deviations are represented as error bars.
2.8. Drug release kinetics
To investigate the preliminary kinetics of DX release from the microspheres, the release data were fit to four basic kinetic models; namely, zero order, first order, Higuchi and Koresemeyer–Peppas equations. These equations are given by:| | |
lnQt = lnQ0 − k1t
| (4) |
here, Mt/M∞ is the fractional drug release at time t; Q0 is the initial amount of drug loaded in the microspheres; Qt is the amount of drug at time t; k0, k1, kH, kKP are the respective kinetic rate constants for the zero order, first order, Higuchi and Korsemeyer–Peppas equations and n is the diffusional exponent indicative of drug transport mechanism and depends on the geometry of the releasing device. For spheres, n = 0.43 signifies Fickian diffusion, n = 0.85 suggests Case II transport which is associated with the relaxational process of the polymers occurring due to water imbibition and for n lying between 0.43 and 0.85 the release mechanism is anomalous in nature where both Fickian and relaxational phenomena contribute to the drug release.39,40 These equations are valid only for the first 60% of drug release.41
2.9. In vitro cytotoxicity assay
The microspheres were first sterilized at 15 lb in−2 steam pressure and 121 °C for 1 h and employed for the cytotoxicity assay using L929NCCS fibroblast cell line (Pune, India). The cell growth was performed on a 24 well tissue culture plate in a controlled atmosphere (5% CO2 at 37 °C) using a cell culture medium of Dulbecco's Modified Eagle's Medium (DMEM, Hi-Media, India) supplemented with 10% Fetal Bovine Serum (FBS, Hi-Media, India) and penicillin–streptomycin antibiotic solution (Hi-Media, India). Ninety percent confluent monolayers of cultured cells were harvested by trypsinization (0.25% Trypsin and 0.02% EDTA, Hi-Media, India) and 1 mL of 1 × 105 cells per mL was seeded in each well. The culture plate was then incubated for 48 h in the CO2 incubator at 37 °C. Cytotoxicity of the microspheres was studied by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] colorimetric assay (Sigma-Aldrich).42
3. Results and discussion
3.1. Inclusion of DX in CD
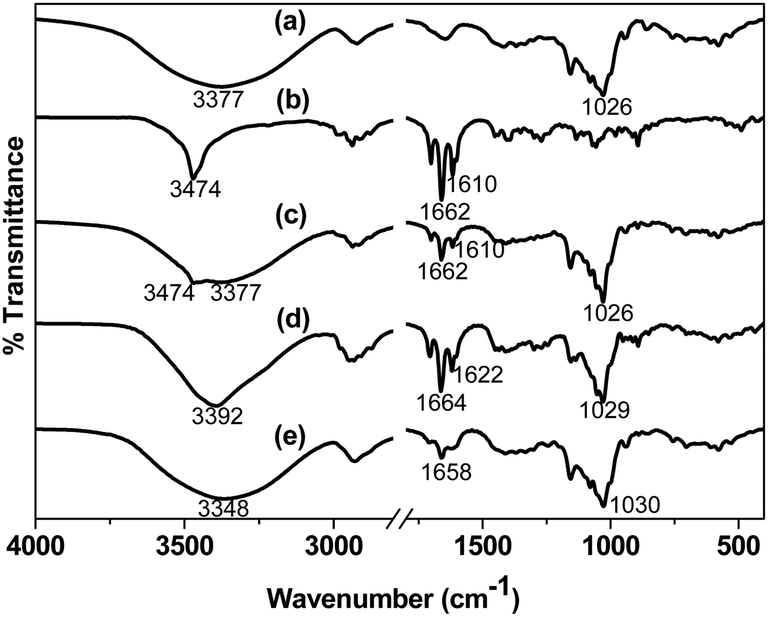
FTIR analysis. Fig. 1 shows the FTIR spectra of CD, DX, PM, CP and FD. The FTIR spectrum of CD (Fig. 1a) shows a broad band with a maximum around 3377 cm−1 due to hydroxyl group stretching, an absorption band at 2920 cm−1 attributed to the C–H stretching and a band at 1645 cm−1 assigned to the bending vibrations of O–H bonds in COH groups and/or in water molecules. The key DX peaks (Fig. 1b) are observed at 1662 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration) and 1610 cm−1 (CC vibration). PM exhibited a spectrum corresponding to the superposition of parent components (Fig. 1c). However the FTIR spectrum of both the inclusion products (Fig. 1d and e) showed shifts in the characteristics hydroxyl and carbonyl absorption positions. A shift in the hydroxyl absorption position from 3377 cm−1 to 3392 cm−1 and 3348 cm−1 in the spectral profiles of CP and FD respectively was evident. Similarly the carbonyl absorption in CP and FD were observed at 1664 and 1658 cm−1 respectively. These spectral shifts in the CP and FD indicated the encapsulation of DX in CD cavity due to the formation of inclusion complexes.43,44
O vibration) and 1610 cm−1 (CC vibration). PM exhibited a spectrum corresponding to the superposition of parent components (Fig. 1c). However the FTIR spectrum of both the inclusion products (Fig. 1d and e) showed shifts in the characteristics hydroxyl and carbonyl absorption positions. A shift in the hydroxyl absorption position from 3377 cm−1 to 3392 cm−1 and 3348 cm−1 in the spectral profiles of CP and FD respectively was evident. Similarly the carbonyl absorption in CP and FD were observed at 1664 and 1658 cm−1 respectively. These spectral shifts in the CP and FD indicated the encapsulation of DX in CD cavity due to the formation of inclusion complexes.43,44
 |
| | Fig. 1 FTIR spectra of (a) CD, (b) DX, (c) PM, (d) CP and (e) FD. | |
XRD analysis. X-ray diffraction has been employed as one of the useful tools to judge drug-CD complexation. The diffraction profile of the inclusion complex is generally different from those of the individual components.45 Fig. 2 presents the wide-angle X-ray diffraction profiles of CD, DX, PM and the inclusion complexes. The CD (Fig. 2a) and DX (Fig. 2b) diffractograms display a series of intense peaks indicating their crystalline nature. The diffraction profile of the PM (Fig. 2c) revealed the features of both CD and DX which suggests that no new crystal has been formed. On the contrary, the diffractogram of CP and FD exhibited completely different features relative to the parent components. CP (Fig. 2d) presented sharp peaks which propose that highly crystalline new crystals of the inclusion complex have been formed. The diffraction profile of FD (Fig. 2e) showed a somewhat amorphous nature of these particles in comparison to CP particles. It is often seen that the freeze-drying method of inclusion complex preparation results in amorphization of drug.46
 |
| | Fig. 2 XRD profiles of (a) CD, (b) DX, (c) PM, (d) CP and (e) FD. | |
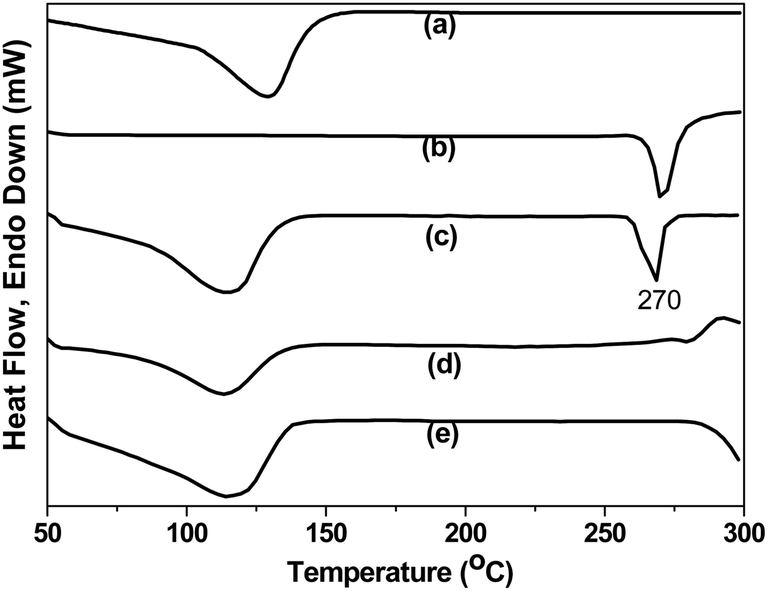
DSC analysis. When guest molecules are embedded in CD cavities; their melting, boiling or sublimating points generally shift to different temperatures or disappear completely.47 The DSC thermograms of CD, DX, PM, CP and FD are shown in Fig. 3. The DSC curve of CD (Fig. 3a) showed a broad endothermic effect around 120 °C which is associated with its dehydration process whereas the thermogram of DX (Fig. 3b) was typical of a crystalline anhydrous substance with a sharp endotherm around 270 °C indicating its melting point. For the PM (Fig. 3c), the endotherms corresponding to the dehydration of CD and DX melting were observed. The DSC curves for CP and FD (Fig. 3d and e respectively) revealed the disappearance of the DX melting peak and only the peak from CD was observed. These results signified the inclusion of DX in CD cavity in the CP and FD.
 |
| | Fig. 3 DSC thermograms of (a) CD, (b) DX, (c) PM, (d) CP and (e) FD. | |
SEM analysis. The SEM micrographs of CD, DX, PM, CP and FD are shown in Fig. 4. CD (Fig. 4a) crystallizes in a larger polyhedral form48 while DX (Fig. 4b) appears as crystalline particles with smaller dimensions. The micrograph of PM (Fig. 4c) presents the crystals of both the parent components. However the crystals of the CP (Fig. 4d) appear as larger blocks which are much different from the sizes and shapes of CD and DX. The FD (Fig. 4e) gave rise to particles with amorphous nature, unlike the morphologies of CD and DX. This agrees with the XRD studies which had shown the amorphous nature for the freeze dried product. Thus the SEM analyses clearly point towards the formation of new entities by the co-precipitation and freeze-drying processes due to the formation of the inclusion complex of DX with CD.50
 |
| | Fig. 4 SEM Micrographs of (a) CD, (b) DX, (c) PM, (d) CP and (e) FD. | |
NMR analysis. Direct evidence for the formation of the inclusion complex can be obtained from 1H NMR studies.49 The 1H NMR spectra for CD, FD and CP are shown in Fig. 5.
 |
| | Fig. 5 1H NMR spectra of (a) CD, (b) FD and (c) CP in D2O at 298 K. | |
The hollow cone topology of CD with H3 and H5 being the inner protons is well established. The hydrophobic guests are included in the toroidal cavity of CD thereby affecting the inner protons. Thus the chemical shift variations in the positions of H3 and H5 of the CD and inclusion complex reflect the formation of an inclusion complex between them. According to Greatbanks and Pickford,50 when [Δ(δH3)] ≤ [Δ(δH5)], then the guest is deeply inside the CD cavity i.e. total inclusion occurs; and if [Δ(δH3)] > [Δ(δH5)] it indicates partial inclusion. The values of chemical shifts, δ, for the protons of CD and those of the inclusion complexes are listed in Table 1.
Table 1 δ and (Δδ) of protons in CD, FD and CP
| Proton |
δCD |
δFD |
Δ(δCD and δFD) |
δCP |
Δ(δCD and δCP) |
| H-1 |
4.982 |
4.880 |
0.102 |
4.891 |
0.091 |
| H-2 |
3.561 |
3.468 |
0.093 |
3.469 |
0.092 |
| H-3 |
3.874 |
3.737 |
0.137 |
3.782 |
0.092 |
| H-4 |
3.496 |
3.405 |
0.091 |
3.408 |
0.088 |
| H-5 |
3.761 |
3.604 |
0.157 |
3.669 |
0.092 |
| H-6 |
3.790 |
3.693 |
0.097 |
3.697 |
0.093 |
From Table 1, it is noteworthy that for both FD and CP, total inclusion of drug in CD cavity is indicated. Molecular dynamics study has also revealed similar possibility of total inclusion of dexamethasone acetate in CD cavity.45 The magnitudes of chemical shifts are relatively higher in case of FD as compared to that in CP, which points towards a somewhat stronger interaction between the drug and CD in FD than in CP complexes. Inclusion complexes prepared by freeze-dried method are often known to be physically more stable than co-precipitated products.51
3.2. Characterization of microspheres
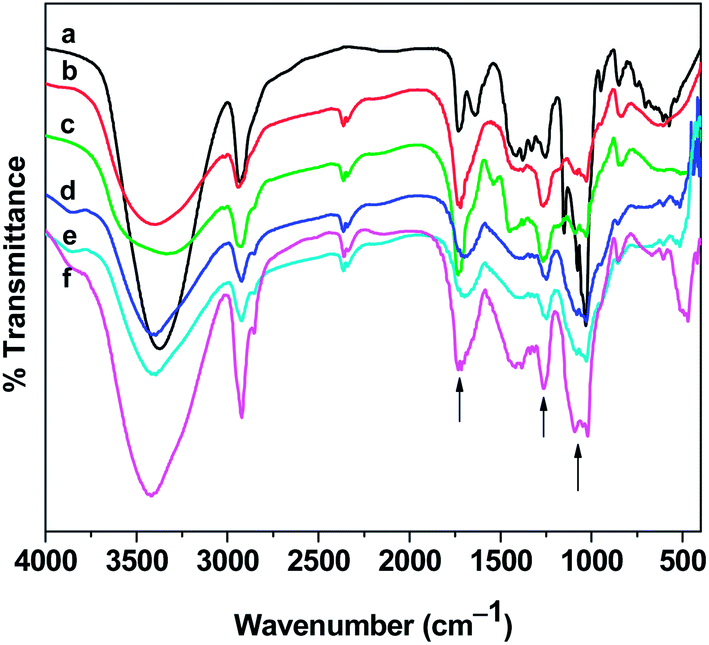
FTIR analysis. The FTIR spectrum of pure PVA shows a large band at around 3400 cm−1 due to hydroxyl stretching (Fig. 6). The C–H stretching from alkyl group regions is observed at around 2941 cm−1 and the peak at 1740 cm−1 is attributed to the CO stretching. The peak at 1151 cm−1 is associated with the crystalline nature of PVA. In the spectra of microspheres, the carbonyl stretching and the region between 1020–1080 cm−1 have been modified indicating the formation of acetal ring by the cross linking reaction between the hydroxyl groups of PVA and aldehydic groups of PAA.52,53 The peak at 1252 cm−1 which is due to C–O stretching vibrations of PAA is enhanced in the spectra of microspheres. This evidenced the incorporation of PAA in the matrices of the synthesized microspheres. In addition, the decrease in intensity in the 1151 cm−1 peak in the microspheres indicates a decrease in crystallinity.
 |
| | Fig. 6 FTIR spectra of (a) pure PVA (b) MS1 (c) MS2 (d) MS3 (e) MS4 and (f) MS5 microspheres. | |
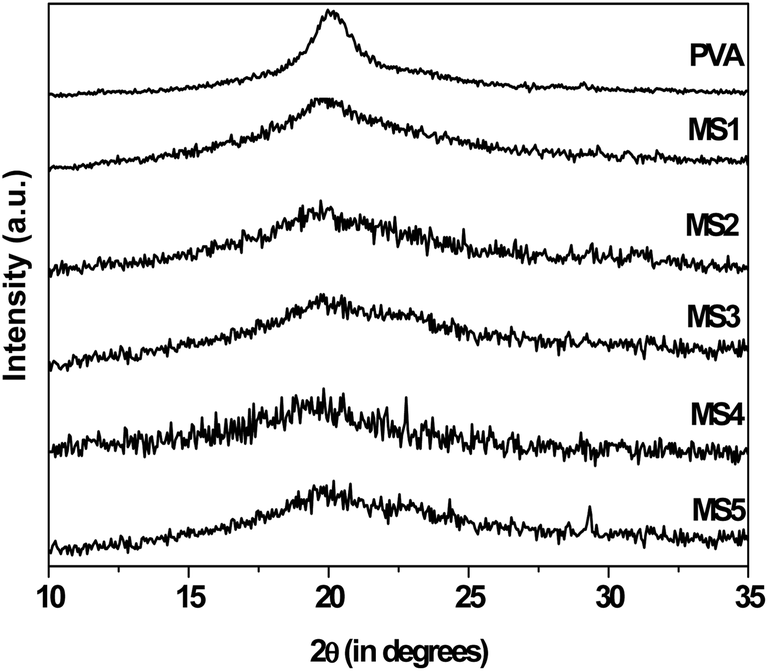
XRD analysis. All samples show almost similar diffraction pattern, a broad peak around 19.7° (Fig. 7). Pure PVA hydrogel shows a peak around diffraction angle (2θ) of 20°, which is associated with the crystalline phase of PVA.54 The relative broadness and decreased intensity observed in the diffraction profiles of the microspheres indicates a decrease in crystallinity in the samples, which might be because of the interpolymer interaction and cross linking of PVA that prevents the PVA chains from self-associating and crystallising. In MS2, the peaks for pure DX are absent and only the peaks for the polymer can be seen. This confirms the molecular dispersion of DX in the polymer matrix and the absence of drug crystallinity. The diffraction profiles of MS3, MS4 and MS5 exhibited more or less similar features and did not show any characteristic CD or DX or inclusion complex peaks. This indicated that the physical mixture and the inclusion complexes are properly blended with the hydrogel matrix.
 |
| | Fig. 7 XRD profiles of pure PVA and MS1, MS2, MS3, MS4 and MS5 microspheres. | |
Morphology analysis. Fig. 8a–e show the SEM images of MS1, MS2, MS3, MS4 and MS5 microspheres. And Fig. 8f–j presents the SEM images of single MS1, MS2, MS3, MS4 and MS5 microsphere. The microspheres are found to be polydispersed in size. They are spherical in shape and formed without any agglomerations. The mean sizes of the microspheres were calculated by considering the average sizes of fifty microspheres of each sample. The mean sizes were found to be 131 ± 23 μm (MS1), 139 ± 24 μm (MS2), 140 ± 22 μm (MS3), 139 ± 21 μm (MS4) and 140 ± 21 μm (MS5). There was no significant difference observed in the morphology of the drug free and drug loaded microspheres. This indicates that the presence of DX or PM or inclusion complex did not have any remarkable effect on the morphology or the size of the microspheres.
 |
| | Fig. 8 SEM Micrographs of (a) MS1, (b) MS2, (c) MS3, (d) MS4, (e) MS5 microspheres at 150× magnification and single (f) MS1, (g) MS2, (h) MS3, (i) MS4, (j) MS5 microspheres at 2000× magnification. | |
3.3. Drug loading efficiency
The drug loading percentages of the MS2, MS3, MS4 and MS5 microspheres are found to be 11.5 ± 0.5, 12.1 ± 0.7, 15.3 ± 0.8 and 17.2 ± 0.7% respectively. The drug loading efficacies of the microspheres demonstrated an increase with the addition of the inclusion complexes of the drug in comparison to the free drug. The MS5 microspheres exhibited highest drug loading capacity probably due to better inclusion complex formation by the freeze-drying method.
3.4. Swelling studies
The swelling studies of the MS1 microspheres in gastric and intestinal pH conditions (pH 1.2 and pH 7.4 respectively) revealed the dependence of the swelling on the pH of external medium. The microspheres exhibited higher swelling at neutral pH as compared to that at acidic pH (Fig. 9). The pH-sensitivity of these microspheres is mainly attributed to the presence of the carboxylic acid group in PAA which is a weak acid with an intrinsic pKa of around 4.28. At pH 1.2, the ionization of carboxylic groups is suppressed and there can be hydrogen bond interaction between the two polymers which reduces the flexibility of the polymer chains. Thus the swelling capacity is lowered. As the pH of the external medium rises above 4.28 (at pH 7.4), the carboxylic groups within the network tend to ionize as a result the inter-polymeric repulsion increases thus increasing the free volume in the polymer matrix which in turn increases the swelling ratio. Additionally, the negative charge in the polymer matrix drives the flow of cations into the hydrogel as a result of which the ionic swelling pressure increases resulting in an increase in swelling.55–57
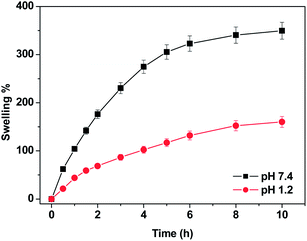 |
| | Fig. 9 Swelling profiles of MS1 microspheres at pH 7.4 and pH 1.2. | |
3.5. In vitro drug release studies
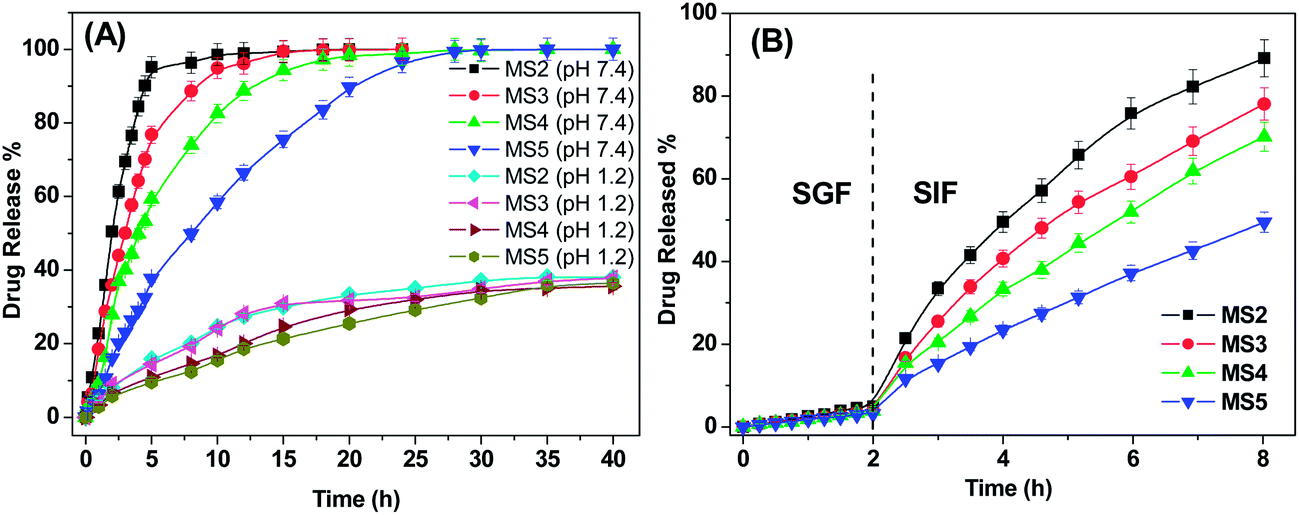
Fig. 10A demonstrates the release profiles of DX from MS2, MS3, MS4 and MS5 microspheres at pH 7.4 and pH 1.2 at 37 °C. As evident, a pronounced difference is observed in the release rates of DX at pH 1.2 and pH 7.4. At pH 1.2, the amounts of DX released from all microspheres were much lower than the release amounts at pH 7.4, even upon prolonged exposure to the releasing medium. This can be attributed to the difference in the extent of swelling of these hydrogels in the above two pH conditions.
 |
| | Fig. 10 Drug release profiles of MS2, MS3, MS4 and MS5 microspheres (A) in pH 7.4, pH 1.2 and (B) in SGF and SIF at 37 °C. | |
At pH 7.4 the rate of release of DX from these hydrogels follows the order: MS2 > MS3 > MS4 > MS5. DX release from the MS2 microspheres was found to be very rapid and almost 80% of the drug has been released in the initial 6 h. However, the release was much prolonged from the MS4 and MS5 microspheres. The release of a drug from a polymer matrix is generally governed by the combined effects of network swelling, polymer relaxation and diffusion of drug from the hydrogel matrix. The observed differences in the release patterns of these microspheres can be rationalised by taking into consideration the physical state of the drug in the hydrogel matrices. The drug release from MS2 microspheres occurs fast due to the easy diffusion of free drug from the hydrogel network to the releasing medium as the hydrogel swells. Whereas in the other three microspheres, the presence of CD in the hydrogel network greatly directs the drug release profiles. For the MS3 microspheres, though the drug release is driven by diffusion, the presence of CD in the hydrogel matrix plays a pivotal role in the achievement of relatively slower drug release rate as compared to that from MS2. Similar decrease in release rate has also been seen for the release of salicylic acid from CD blended PVA hydrogel as compared to pure PVA hydrogel.31 This can be explained by considering the following possibilities: (i) a few of the drug molecules getting complexed with the CD during the hydrogel synthesis and/or (ii) presence of H-bonding between CD and the polymer matrix influencing the polymer relaxation which in turn effects the drug release rate. For the drug release from MS4 and MS5 microspheres, apart from all the above mentioned factors, one major aspect that needs to be considered is the strong binding of DX with CD (Ka = 700 ± 40 M−1, determined by HPLC studies).58 Thus a sustained release of DX is observed from these microspheres in contrast to the burst type release from MS2 microspheres.
A pronounced difference in the release rates from MS4 and MS5 microspheres is observed. The drug release from MS5 was much more controlled and continued for longer time than MS4 microspheres. These two microspheres are very similar in all aspects except for the type of inclusion complex. Thus, the observed difference in the drug release kinetics between MS4 and MS5 suggests towards a strong influence of the method of preparation of the inclusion complex on the drug release kinetics from the hydrogel matrix.
In order to imitate the conditions of the gastrointestinal tract (GIT), DX release from the microspheres was studied in SGF and SIF; the microspheres were immersed in SGF for 2 hours and then transferred to SIF and the drug release was monitored. Fig. 10B depicts the release pattern of DX from the four microspheres in SGF and SIF environments. Approximately 5% of DX is released during the initial 2 h in SGF. However, when the microspheres were transferred to SIF, the rate of DX release increased significantly for all the four hydrogels. The release rate in the SIF was found to follow the same order as observed before i.e. MS2 > MS3 > MS4 > MS5. This release profile of dexamethasone fulfils the requirements of US Pharmacopeia for oral drug delivery to the lower part of the GIT (USP XXIV).59
Thus the synthesized microspheres could be effectively employed for the oral delivery of dexamethasone and their pH sensitivity could be exploited for the delivery to the intestine. However, in order to achieve a controlled release of drug the MS4 and MS5 microspheres are found to be more suitable. And MS5 is found to be the best system for achieving the slow release of DX. Thus, it can be proposed that the adverse side-effects of DX could be minimized by using the MS5 microspheres as delivery vehicles since they provide controlled release of DX over a prolonged period.
3.6. Drug release kinetics
The release data have been fitted to different empirical mathematical equations using Origin 7 software (Origin Lab Corporation) and the correlation coefficient R2 are shown in Table 2. The drug release data for all the microspheres showed the best fit for the Korsemeyer–Peppas equation. The diffusion exponent value ‘n’ was found to be in the range of 0.76 to 0.84 for MS3, MS4 and MS5 microspheres indicating the anomalous nature of drug transport mechanism, which is the superimposition of diffusion-controlled and swelling controlled drug release. For MS2 the ‘n’ value is found to be 0.85 implying the Case II type drug transport, where the relaxation process of the polymer matrix upon water imbibition into the system is the rate-controlling step. This is further supported by the fact that the release data from MS2 fit quite well to the zero-order kinetics (R2 = 0.996).
Table 2 Fitting parameters (R2) for the release data estimated from different mathematical equations
| Formulation |
Zero order |
First order |
Higuchi |
Korsemeyer–Peppas |
| n |
R2 |
| MS2 |
0.996 |
0.992 |
0.822 |
0.85 |
0.998 |
| MS3 |
0.989 |
0.987 |
0.876 |
0.84 |
0.998 |
| MS4 |
0.979 |
0.985 |
0.849 |
0.76 |
0.992 |
| MS5 |
0.958 |
0.988 |
0.881 |
0.80 |
0.996 |
3.7. Cytotoxicity assay
In order to ensure the biocompatibility of the synthesized microspheres, cytotoxicity assay was performed by MTT colorimetric technique. As shown in Fig. 11, direct contact between L929 cells and MS1 microspheres did not reveal any adverse effect. This suggested the compatibility of the synthesized microspheres with the living tissues thus validating these as possible drug delivery systems.
 |
| | Fig. 11 Optical micrographs of L-929 cells cultured after 48 h incubation with MS1 microspheres. | |
4. Conclusions
pH-sensitive PVA–PAA microspheres cross linked with glutaraldehyde were synthesized and studied for the delivery of the common anti-inflammatory and immunosuppressant drug dexamethasone. To regulate the release rate of DX, preformed solid inclusion complex of DX with CD was added into the hydrogel microspheres. In order to examine the effect of the method of preparation of inclusion complex on the release kinetics, the inclusion complex was prepared by two different methods: the co-precipitation and freeze-drying method. The swelling characteristics indicated higher swelling in neutral pH than in acidic pH. At pH 1.2, the amounts of DX released from all microspheres were much lower than the release amounts at pH 7.4. At pH 7.4 the rate of release of DX follows the order: MS2 > MS3 > MS4 > MS5. Slowest release was observed from the MS5 microspheres which contained the freeze-dried inclusion complex. The drug release was also studied in SGF and SIF at 37 °C; approximately 5% of DX released during the initial 2 h in SGF but upon transfer to SIF, the rate of DX release increased significantly. Thus the synthesized microspheres could be effectively employed for the controlled delivery of dexamethasone and their pH sensitivity could be exploited for the delivery to the intestine. Moreover, the compatibility of the synthesized microspheres with the living tissues further validates them as promising drug delivery systems. MS5 containing the freeze-dried inclusion complex was found to be the best of the lot for achieving a good control on the delivery of DX. Thus, it can be proposed that the adverse side-effects of DX could be minimized by using the MS5 microspheres as delivery vehicles since they provide controlled release of DX over a prolonged period.
Acknowledgements
The authors thank the Department of Science and Technology, India for the financial support.
References
- R. V. Kulkarni, R. Boppana, G. K. Mohan, S. Mutalik and N. V. Kalyane, J. Colloid Interface Sci., 2012, 367, 509–517 CrossRef CAS PubMed
 .
. - L. Ma, M. Liu, H. Liu, J. Chen, C. Gao and D. Cui, Polym. Adv. Technol., 2010, 21, 348–355 CAS .
- A. Vats and K. Pathak, Expert Opin. Drug Delivery, 2013, 10, 545–557 CrossRef CAS PubMed .
- G. Van den Mooter, Expert Opin. Drug Delivery, 2006, 3, 111–125 CrossRef CAS PubMed .
- M. K. Chourasia and S. K. Jain, J. Pharm. Pharm. Sci., 2003, 6, 33–40 CAS .
- S. Das and U. Subuddhi, Ind. Eng. Chem. Res., 2013, 52, 14192–14200 CrossRef CAS .
- Q. Wang, J. Zhang and A. Wang, Carbohydr. Polym., 2009, 78, 731–737 CrossRef CAS PubMed .
- Y. S. R. Krishnaiah, P. V. Raju, B. D. Kumar, P. Bhaskar and V. Satyanarayana, J. Controlled Release, 2001, 77, 87–95 CrossRef CAS .
- B. Singh and V. Sharma, Int. J. Pharm., 2010, 389, 94–106 CrossRef CAS PubMed .
- M. Liu, L. Wang, H. Su, H. Cao and T. Tan, Polym. Bull., 2013, 70, 2815–2827 CrossRef CAS .
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS PubMed .
- B. Gyarmati, A. Nemethy and A. Szilagyi, RSC Adv., 2014, 4, 8764–8771 RSC .
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS .
- K. M. Huh, H. C. Kang, Y. J. Lee and Y. H. Bae, Macromol. Res., 2012, 20, 224–233 CrossRef CAS PubMed .
- M. C. I. M. Amin, N. Ahmed, N. Halib and I. Ahmed, Carbohydr. Polym., 2012, 88, 465–473 CrossRef PubMed .
- M. Cheddadi, E. Lopez-Cabarcos, K. Slowing, E. Barcia and A. Fernandez-Carballido, Int. J. Pharm., 2011, 413, 126–133 CrossRef CAS PubMed .
- H. A. Abd El-Rehim, E. A. Hegazy, F. H. Khalil and N. A. Hamed, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 254, 105–112 CrossRef CAS PubMed .
- S. Das and U. Subuddhi, J. Appl. Polym. Sci. DOI:10.1002/app.40318 .
- B. Gajra, S. S. Pandya, G. Vidyasagar, H. Rabari, R. R. Dedania and S. Rao, Int. J. Pharm. Res., 2012, 4, 20–26 CAS .
- P. Basak and B. Adhikari, J. Mater. Sci.: Mater. Med., 2009, 20, S137–S146 CrossRef CAS PubMed .
- D. Ray, P. Sunny Gils, G. P. Mohanta, R. Manavalan and P. K. Sahoo, J. Appl. Polym. Sci., 2010, 116, 959–968 CAS .
- I. Manavi-Tehrani, M. Rabiee, M. Parviz, M. R. Tahiri and Z. Fahimi, Macromol. Symp., 2010, 296, 457–465 CrossRef CAS .
- H. Byun, B. Hong, S. Y. Nam, S. Y. Jung, J. W. Rhim, S. B. Lee and G. Y. Moon, Macromol. Res., 2008, 16, 189–193 CrossRef CAS .
- H. S. Shin, S. Y. Kim and Y. M. Lee, J. Appl. Polym. Sci., 1997, 65, 685–693 CrossRef CAS .
- A. D. McLeod, D. R. friend and T. N. Tozer, J. Pharm. Sci., 1994, 83, 1284–1288 CrossRef CAS .
- R. N. Fedorak, B. Haeberlin, L. R. Empey, N. Cui, H. N. Ill, L. D. Jewell and D. R. Friend, Gastroenterology, 1995, 108, 1688–1699 CrossRef CAS .
- Y. Mao, G. Triantafillou, E. Hertlein, W. Towns, M. Stefanovski, X. Mo, D. Jarjoura, M. Phelps, G. Marcucci, L. J. Lee, D. M. Goldenberg, R. J. Lee, J. C. Byrd and N. Muthusamy, Clin. Cancer Res., 2013, 19, 347–356 CrossRef CAS PubMed .
- M. Lasa, M. Brook, J. Saklatvala and A. R. Clark, Mol. Cell. Biol., 2001, 21, 771–780 CrossRef CAS PubMed .
- R. Cavalli, F. Trotta and W. Tumiatti, J. Inclusion Phenom. Mol. Recognit. Chem., 2006, 56, 209–213 CrossRef CAS .
- D. C. Bibby, N. M. Davies and I. G. Tucker, Int. J. Pharm., 2000, 197, 1–11 CrossRef CAS .
- K. Sreenivasan, J. Appl. Polym. Sci., 1997, 65, 1829–1832 CrossRef CAS .
- V. Mohit, G. Harshal, D. Neha, K. Vilasrao and H. Rajashree, J. Inclusion Phenom. Mol. Recognit. Chem., 2010, 67, 39–47 CrossRef CAS .
- J.-Y. Tsao, H.-H. Tsai, C.-P. Wu, P.-Y. Lin, S.-Y. Su, L.-D. Chen, F.-J. Tsai and Y. Tsai, Int. J. Pharm., 2010, 402, 123–128 CrossRef CAS PubMed .
- P. J. Salustio, G. Feio, J. L. Figueirinhas, J. F. Pinto and H. M. Cabral Marques, Eur. J. Pharm. Biopharm., 2009, 71, 377–386 CrossRef CAS PubMed .
- M. D. Kurkuri and T. M. Aminabhavi, J. Controlled Release, 2004, 96, 9–20 CrossRef CAS PubMed .
- G. Mino and S. Kaizerman, J. Appl. Polym. Sci., 1958, 31, 242–243 CrossRef .
- S. C. Jana, S. Maiti and S. Biswas, J. Appl. Polym. Sci., 2000, 78, 1586–1590 CrossRef CAS .
- S. C. Angadi, L. S. Manjeshwar and T. M. Aminabhavi, Ind. Eng. Chem. Res., 2011, 50, 4504–4514 CrossRef CAS .
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS .
- C.-C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS PubMed .
- L. Serra, J. Domenech and N. A. Peppas, Biomaterials, 2006, 27, 5440–5451 CrossRef CAS PubMed .
- L. E. Smith, S. Rimmer and S. MacNeil, Biomaterials, 2006, 27, 2806–2812 CrossRef CAS PubMed .
- R. F. L. Vianna, M. V. L. B. Bentley, G. Ribeiro, F. S. Carvalho, A. F. Neto, D. C. R. de Oliveira and J. H. Collett, Int. J. Pharm., 1998, 167, 205–213 CrossRef CAS .
- M. M. Doile, K. A. Fortunato, I. C. Schmucker, S. K. Schucko, M. A. S. Silva and P. O. Rodrigues, AAPS PharmSciTech, 2008, 9, 314–321 CrossRef CAS PubMed .
- Q. Zhou, X. Wei, W. Dou, G. Chou and Z. Wang, Carbohydr. Polym., 2013, 95, 733–739 CrossRef CAS PubMed .
- M. K. Riekes, M. P. Tagliari, A. Granada, G. Kuminek, M. A. S. Silva and H. K. Stulzer, Mater. Sci. Eng., 2007, 30, 1008–1013 CrossRef PubMed .
- H. C. Marques, J. Hadgraft and I. Kellaway, Int. J. Pharm., 1990, 63, 259–266 CrossRef .
- L.-J. Yang, W. Chen, S.-X. Ma, Y.-T. Gao, R. Huang, S.-J. Yan and J. Lin, Carbohydr. Polym., 2011, 85, 629–637 CrossRef CAS PubMed .
- H. J. Schneider, F. Hacket and V. Rudigger, Chem. Rev., 1998, 98, 1755–1785 CrossRef CAS PubMed .
- D. Greatbanks and R. Pickford, Magn. Reson. Chem., 1987, 25, 208–213 CrossRef CAS .
- G. Zingone and F. Rubessa, Int. J. Pharm., 2005, 291, 3–10 CrossRef CAS PubMed .
- L. Wang, J. Li, Y. Lin and C. Chen, J. Membr. Sci., 2007, 305, 238–246 CrossRef CAS PubMed .
- Y. Lu, D. Wang, T. Li, X. Zhao, Y. Cao, H. Yang and Y. Y. Duan, Biomaterials, 2009, 30, 4143–4151 CrossRef CAS PubMed .
- P. R. Hari and K. Sreenivasan, J. Appl. Polym. Sci., 2001, 82, 143–149 CrossRef CAS .
- J. Byun, Y. M. Lee and C.-S. Cho, J. Appl. Polym. Sci., 1996, 61, 697–702 CrossRef CAS .
- J. Fei, Z. Zhang, L. Zhong and L. Gu, J. Appl. Polym. Sci., 2002, 85, 2423–2430 CrossRef CAS .
- S. M. M. Quintero, R. V. Ponce, F. M. Cremona, A. L. C. Triques, A. R. d'Almeida and A. M. B. Braga, Polymer, 2010, 51, 953–958 CrossRef CAS PubMed .
- N. Sadlej-Sosnowska, J. Inclusion Phenom. Mol. Recognit. Chem., 1997, 27, 31–40 CrossRef CAS .
- A. Islam and T. Yasin, Carbohydr. Polym., 2012, 88, 1055–1060 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.