
Phloroglucinol-encapsulated starch biopolymer: preparation, antioxidant and cytotoxic effects on HepG2 liver cancer cell lines
Abstract
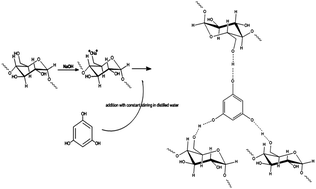
Phloroglucinol-encapsulated starch biopolymer was prepared by graft polymerization in the presence of sodium hydroxide as catalyst. The biopolymer was characterized by Fourier Infrared spectroscopy (FT-IR), scanning electron microscopy (SEM), X-ray diffraction (XRD), dynamic light scattering and 1H nuclear magnetic resonance spectroscopy (1H NMR). The in vitro quantification of the biopolymer revealed that phloroglucinol was encapsulated by soluble starch at a ratio of 3 : 1. It is also observed that the antioxidant potential of the biopolymer were significantly high in terms of concentration. The cytotoxic assay showed that the biopolymer is effective in killing HepG2 liver cancer cells lines in a dose-dependent manner with morphological evidence. To conclude, the comprehensive applications of phloroglucinol-encapsulated starch biopolymer agrees with green chemistry principles to satisfy future needs in the field of medical and pharmaceutical industry.
 Please wait while we load your content...
Please wait while we load your content...

