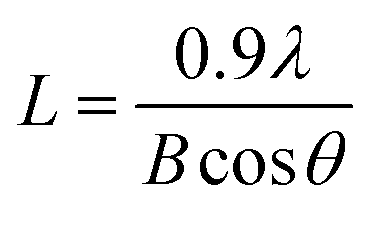DOI:
10.1039/C4RA02542C
(Paper)
RSC Adv., 2014,
4, 26140-26148
Platinum-decorated chemically modified reduced graphene oxide–multiwalled carbon nanotube sandwich composite as cathode catalyst for a proton exchange membrane fuel cell
Received
23rd March 2014
, Accepted 21st May 2014
First published on 21st May 2014
Abstract
Graphite oxide (GO) was chemically altered using polyethylene glycol (PEG) to produce functionalized GO (f-GO). An ensemble of reduced f-GO sheets and multiwalled carbon nanotubes (MWNTs), referred to as few-layer graphene–MWNT sandwiches (GCSs), were synthesized by a catalysis-assisted chemical vapor deposition (CCVD) method and explored as the electrocatalyst support material for oxygen reduction reaction (ORR) in a proton exchange membrane fuel cell (PEMFC). Platinum nanoparticles were decorated on the carbon supports by a modified glycol reduction technique. As-prepared electrocatalysts were characterized by Raman spectroscopy, X-ray diffraction and transmission electron microscopy. Electrocatalytic performance was evaluated by cyclic voltammetry and PEMFC fuel cell measurements and compared with a commercially available Pt/C electrocatalyst. The Pt/GCS electrocatalyst gave a maximum PEMFC performance of 495 mW cm−2 at 60 °C temperature. The improvement in the ORR activity was ascribed to the uniform dispersion of Pt nanoparticles with an optimal particle size (∼3.5 nm) over a well-organized conducting catalyst support.
Introduction
In the present day scenario of industrial revolution, the high demand of energy sources has led to enhanced consumption of fossil fuels, resulting in a large number of atmospheric changes such as global warming, an increase in greenhouse gases and acid rain. To meet this challenge at low cost, much research is directed at finding various renewable and alternative energy sources. Hydrogen, with its highest energy density (141 MJ kg−1), is abundant in nature, and when employed properly, results in almost no pollution compared to the use of fossil fuels. Among different electrochemical energy conversion devices, the fuel cell is very promising.1 Out of various fuel cells, a low temperature-based hydrogen–oxygen (H2–O2) proton exchange membrane fuel cell (PEMFC) has attracted much attention in the scientific world for its advantages. The advantages of PEMFC such as high operating voltage, high efficiency, quick start up, no emission and portability, make it an important and popular energy conversion device for future resources.2–4 However, the slow kinetics of the oxygen reduction reaction (ORR) at the cathode produces a large overpotential, thereby decreasing the cell's performance.5 Platinum (Pt) is the main catalyst used in PEMFC because of its excellent catalytic activity compared with other metals in a harsh acidic environment.6 However, the high cost and limited resources of the Pt catalyst are the main reasons for the non-commercialization of PEMFC. The electrocatalytic activity of Pt depends on several factors, such as the size and shape of Pt particles, nature of the support material and dispersion of Pt over the support.7–9 To reduce Pt content, alloying of Pt or maximum utilization of Pt by reducing the particle size (3–5 nm) along with high dispersion over a proper support material has been used.10–17 A commercially used support, such as carbon black suffers from the disadvantages of corrosion and weak bonding of Pt with the support, leading to aggregation of nanoparticles and decrease in performance. High surface area carbon materials with good electrical conductivity such as graphene and multiwalled carbon nanotubes (MWNTs) have gained a lot of attention over many conventional catalyst support materials for various energy applications.18–24 MWNTs have been reported to be good catalyst support materials for PEMFC applications.21,23 In the recent past, graphene has come forward as a perfect material for various energy applications such as in the Li ion battery, as a supercapacitor and in fuel cells, among others.25–27
Recently, the composite of MWNTs and reduced graphene oxide or few-layer graphene sheets is reported to be one of the ideal supports because of the unique combination of good electrical conductivity and high surface area.28,29 Other than mechanical mixing of these two,29 preparation of the composite can also be done as we reported earlier: relying on the electrostatic interaction between acid functionalized MWNT and poly(diallyldimethylammonium chloride) (PDDA)-treated graphene.30 It has been shown that a uniform distribution of MWNTs can act as conductive spacers between graphene sheets. These techniques, although they provide good distribution of the (graphene–MWNT) composite, require a time-consuming preparation method because of the addition of several steps into the synthesis. In another report, Jyothirmayee et al. studied graphene-wrapped carbon nanotubes as a catalyst support material.31 In this method, the wrapping of graphene sheets around MWNT does not significantly increase the effective surface area of the catalyst support for the dispersion of Pt nanoparticles. Here, we report a simple and easily scalable technique for synthesizing a few-layer graphene–MWNTs sandwich (GCS) structure by the surface passivation of graphene oxide and subsequent chemical vapor deposition (CVD). Among the various reduction methods of Pt decoration over a support material, the polyol reduction method results in the uniform dispersion of particles with an optimal size.32,33 In the polyol reduction method, ethylene glycol (EG) acts both as the surfactant and as the reducing agent. The maximum Pt metal loading over the support material can be obtained using this method to achieve a uniform, narrow-size distribution; it can also be restricted by adjusting the water to EG ratio, the pH of liquid reaction system and impregnation time of Pt precursor to support material. Pt decorated by this method over GCS is further used as a cathode electrocatalyst for PEMFC applications.
Experimental section
Synthesis of electrocatalyst
Graphite oxide (GO) was prepared from graphite powder using a modified Hummer's method.34,35 Briefly, 2 g of graphite powder was added to 46 ml concentrate H2SO4 under stirring in an ice bath, and 1 g NaNO3 and 6 g KMnO4 were added slowly into that solution. This suspension was removed from the ice bath and allowed to come to room temperature. Warm water (280 ml) was added to dilute the solution. H2O2 (3%) was added until the color of the solution changed to bright yellow. This final solution was then filtered and washed several times with warm water. The residue was diluted using water, then the solution was centrifuged and dried at 60 °C. This dried sample (GO) was first exposed to concentrated acid (H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1) to adhere oxygen-containing functional groups to the sample. Polyethylene glycol (PEG) was used to produce surface-passivized GO (f-GO). Briefly, a solution mixture of GO and PEG was ultrasonicated for 1 h and stirred for 3 h. Polymer chains of PEG became attached to the GO sheets by replacing some of the functional groups adsorbed in the previous acid treatment step. The solution was then filtered and washed with copious amounts of DI water to remove any non-bonded PEG on the GO surface. The sample was then vacuum dried to produce f-GO.
:HNO3 = 3:1) to adhere oxygen-containing functional groups to the sample. Polyethylene glycol (PEG) was used to produce surface-passivized GO (f-GO). Briefly, a solution mixture of GO and PEG was ultrasonicated for 1 h and stirred for 3 h. Polymer chains of PEG became attached to the GO sheets by replacing some of the functional groups adsorbed in the previous acid treatment step. The solution was then filtered and washed with copious amounts of DI water to remove any non-bonded PEG on the GO surface. The sample was then vacuum dried to produce f-GO.
This f-GO was mixed with a misch-metal-based alloy (MmNi3) hydride at a 2:1 ratio using a mortar and pestle. A hydrogen decrepitating technique was used to extract the fine particles (2–10 μm) from the ingot of MmNi3. Each alloy hydride particle contains a number of catalytic centers that act as nucleation sites for the growth of MWNTs, as we reported previously.36 The mixture was uniformly blended by grinding for few minutes and then spread over a quartz boat, which was then placed at the centre of a horizontal quartz tube reactor, located inside a furnace. The tube was sealed with end couplings with provision for gas flow. The tube was first flushed with argon gas at room temperature for 15 min. Highly pure hydrogen (99.99%) gas was introduced, and the temperature was raised to 500 °C. In this atmosphere, the oxygen-containing functional groups, such as –OH, –COOH groups, present in the system react rapidly with hydrogen gas. Flames were seen for less than a second as a result of the exothermic violent reaction. This reaction provides adequate energy for the exfoliation of the carbon planes. Both reduction of oxygen functional groups by the formation of water vapor and exfoliation of planes took place simultaneously, leading to the formation of reduced f-GO (Rf-GO) or few-layer graphene sheets. The temperature was further increased to 700 °C, and acetylene gas as the carbon precursor was allowed for 30 min. The precursor gas at this temperature decomposes to release carbon, which first dissolves on the metal surface and then diffuses through the molten alloy. MWNTs were grown on these saturated metal carbide catalytic centers by the well-reported vapor–liquid–solid (VLS) mechanism.37–39 The surface passivation of GO using PEG in this technique prevented the graphene surface from wrapping around the nanotube surfaces, as reported earlier,31 leading to a sandwich-like structure with a uniform distribution of MWNTs between the graphene sheets. The furnace was then switched off and allowed to cool to room temperature in the presence of argon flow. The product was collected after cooling and air-oxidized at 350 °C for 3 h to remove any amorphous carbon present. The sample was collected after cooling to room temperature and described as a few-layer graphene–multiwalled carbon nanotube sandwich (GCS) structure. This as-synthesized material was used as the electrocatalyst support material without further modification. MWNTs were synthesized by the same procedure without the addition of f-GO. For comparison, pristine few-layer graphene sheets (Rf-GO) were obtained from f-GO by performing the previously mentioned procedure, heating up to 500 °C under H2 atmosphere without adding the MmNi3 hydride catalyst.
Platinum decoration on the support material was achieved by modified polyol reduction method. In this method, 100 mg of GCS was dispersed in 100 ml of ethylene glycol (MW = 62.07 g mol−1) solution (EG:water = 2:1) by ultrasonication for 1 h followed by 12 h stirring. 11.38 ml of hexachloroplatinic acid (H2PtCl6·6H2O) was added drop-wise to the above solution and stirred for another 24 h. The loading of platinum on the support material was controlled to 30%. pH of the solution was adjusted to 11 by adding NaOH solution; it was then refluxed at 130 °C for 6 h to ensure the complete reduction of the precursor acid. This product was then washed several times with DI water and dried at 60 °C in a vacuum oven to produce Pt/GCS. Dispersion of Pt over Rf-GO sheets and MWNTs was also carried out by a similar method.
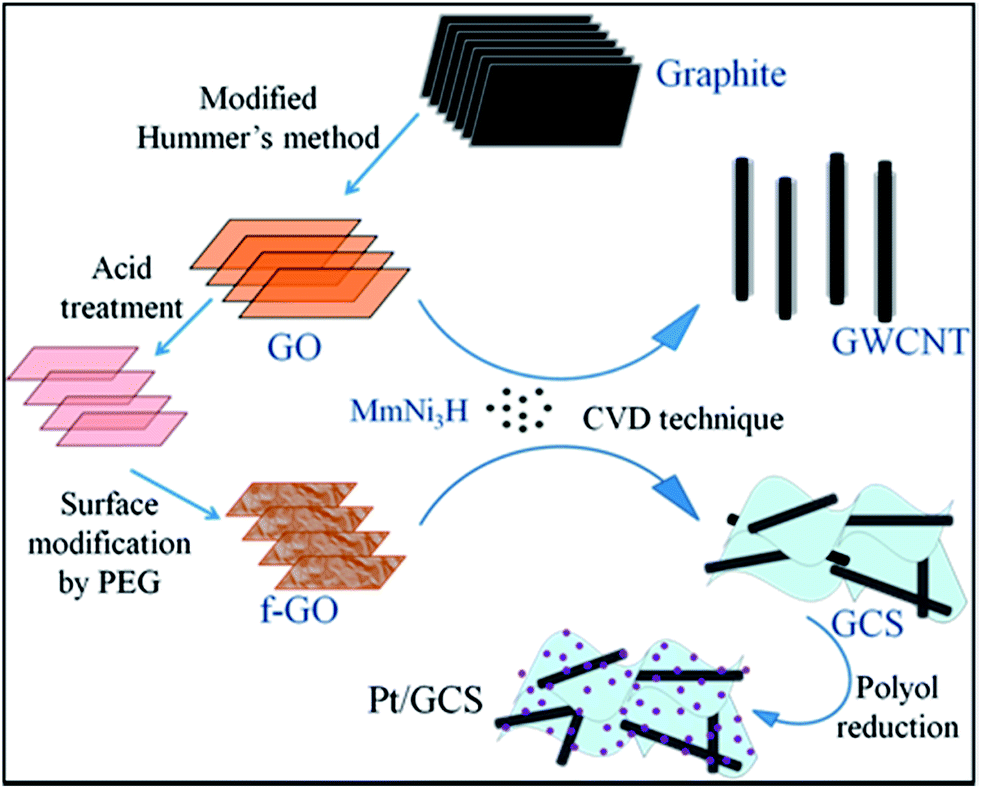
Fig. 1 is a schematic of the procedure for the Pt/GCS electrocatalyst synthesis. GO was prepared from graphite by a modified Hummers method and further acid-treated to break down into a smaller size towards the nano regime, along with the adherence of more oxygen functional groups. PEG was subsequently employed to modify the surfaces of these small-sized GOs. The polar nature of the polymer molecule allows it to intercalate between sheets that causes an expansion in the basal plane spacing, resulting in an easy process of synthesis for less-wrinkled and fewer-layered graphene sheets.40 Water-soluble PEG was used to passivize the surface to produce a sandwich-like structure and not graphene-wrapped MWNTs. This sandwich structure of MWNTs between graphene layers prevents restacking of the sheets, providing more surface area with high electrical conductivity. The high surface area of 2D Rf-GO and 1D MWNT provides the Pt nanoparticles with more anchor sites to attach to the support material. Faster electron transfer was allowed by the presence of highly conductive MWNTs.41
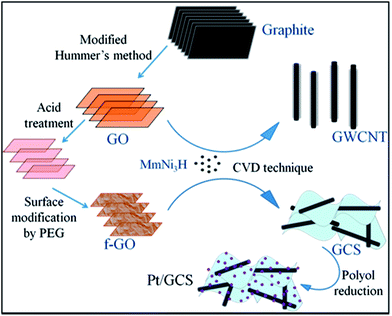 |
| | Fig. 1 Schematic representation of the synthesis of Pt/GCS electrocatalyst in contrast with Pt/wrapped graphene–MWNT. | |
Physical characterizations
X-ray diffraction (XRD) measurements were carried out in a PANalytical X'Pert Pro X-ray diffractometer with a nickel-filtered Cu-Kα radiation source (λ = 0.15406 nm) at 40 kV and 30 mA. The diffraction data was recorded in the range of 5–90° (2θ) with a step size of 0.016°. Raman spectroscopy study using a WiTec Alpha 300 analyzed the vibrational modes of the samples with a 532 nm, Nd:YAG laser as the excitation source. A high resolution transmission electron microscopy instrument (HRTEM, Technai G20 (200 kV)) was used to study the morphology of the synthesized samples. For this purpose, the samples were prepared by dispersion in ethanol by ultrasonication and drop-casting on a perforated carbon-coated 200 mesh copper grid. After drying in ambient atmosphere, the coated copper grids were used to study the texture of the samples.
Electrochemical characterizations
Electrochemical measurements were carried out in a 3-electrode-based CHI 608C instrument using Pt wire as the counter and Ag/AgCl (dipped in 1 M KCl) as the reference electrode in 1 M H2SO4 electrolyte solution at room temperature. A slurry was prepared by ultrasonication of 5 mg of sample mixed with 5 μl of Nafion solution and 200 μl of ethanol for 30 min. The working electrode was prepared by dropping a measured quantity (3 μl) of this prepared slurry in a polished glassy carbon electrode (GCE) substrate of area 0.07 cm2 and dried for 24 h under ambient conditions. Activation scans were performed before taking the final result to check the reproducibility of the voltammograms.
For full cell measurements, the electrodes were prepared by coating the catalyst ink over the carbon cloth gas diffusion layer. The catalyst ink was prepared by ultrasonicating the measured amount of catalyst, 2-propanol and Nafion (5 wt%) solution. The amount of Pt loading was controlled to 0.25 mg cm−2 and 0.5 mg cm−2 for the anode and cathode, respectively. The membrane electrode assembly (MEA) was prepared by sandwiching a pretreated 212 CS Nafion membrane between the anode and cathode by hot pressing at 130 °C for 4 min at 1 ton pressure. The effective electrode area was 11.56 cm2. Each MEA was tested by fitting it between two graphite plates, having the gas-flow condition in a serpentine geometry. The flow of hydrogen and oxygen was controlled to 100 sccm at 90% humidity. A humidifier with inline heating was used to maintain the required water content in MEA. Full cell measurements were carried out by the Teledyne instrument. The MEA was first activated between an open circuit potential condition and high current densities. Measurements were done at three different temperatures: 40 °C, 50 °C and 60 °C. For MEA preparation, Pt/MWNT was used as the anode catalyst for all cathode catalysts: commercial Pt/C, synthesized Pt/Rf-GO and Pt/GCS, designated MEA 1, MEA 2 and MEA 3, respectively.
Results and discussion
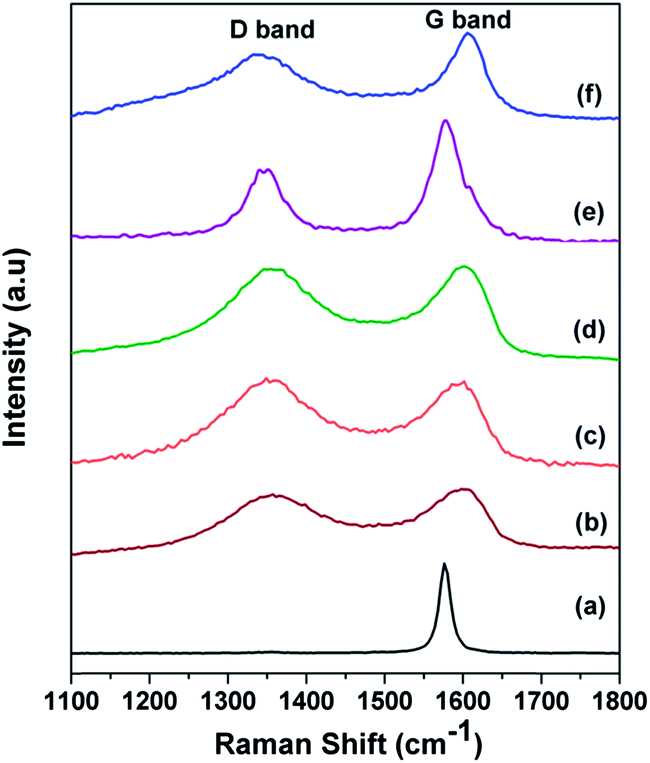
Raman spectra for the graphite, GO, f-GO, Rf-GO, MWNT and GCS are shown in Fig. 2 for the range 1100 to 1800 cm−1. A high-intensity laser beam can impart sufficient amount of energy for the exfoliation of GO locally, and burning of material is also possible at higher intensity; the present Raman measurements were carried out with a low intensity laser. The D band is common to all sp2 hybridized disordered carbon materials; it signifies the degree of disorder. The G band is due to the Raman active E2g mode and also represents the crystallinity of a material; a more intense G peak signifies a more highly crystalline material. The sharp G band and almost negligible D band in Fig. 2a indicate the presence of fewer defects in the graphite.42 For GO, the broadened D band with higher intensity suggests the incorporation of functional defects, and the wide G band suggests a disturbance in crystallinity as compared to graphite (Fig. 2b). A positive shift can be seen for the G band in Fig. 2b from graphite, 1576 cm−1. In Fig. 2c f-GO, the D band is more intense as compared to that of GO, suggesting the enhancement of defects due to the chemical modifications by the incorporation of functional groups and polymer PEG. The G band in Fig. 2c is downshifted, suggesting the possibility of electron transfer from the surface-adhered PEG to GO.43 For Rf-GO, the removal of oxygen functional groups gives a slightly lowered D band as compared to f-GO in Fig. 2d. The sharp G band in Fig. 2e is ascribed to the high degree of crystallinity in MWNTs. The reduction of f-GO and formation of MWNTs in the composite is confirmed by a lower D band and sharp G peak (Fig. 2f). The ratio of the intensities of D band to the G band (ID/IG) for these materials are given in Table 1.
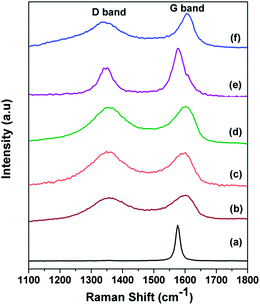 |
| | Fig. 2 Raman spectra of (a) graphite, (b) GO, (c) f-GO, (d) Rf-GO, (e) MWNTs, (f) GCS. | |
Table 1 Raman ID/IG ratio of the carbon materials used in the present work
| Material |
ID/IG ratio |
| Graphite |
0.012 |
| GO |
0.892 |
| f-GO |
0.975 |
| Rf-GO |
0.97 |
| MWNT |
0.53 |
| GCS |
0.77 |
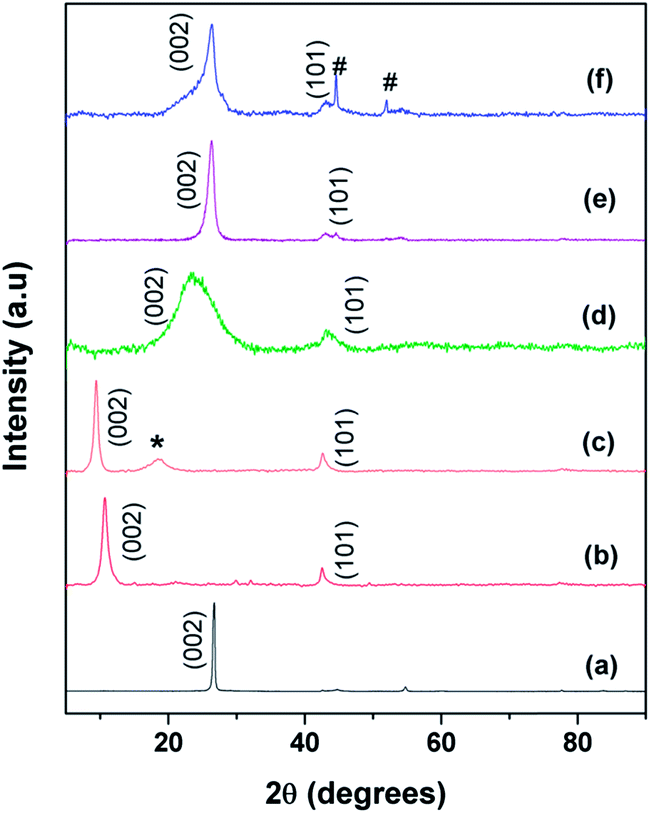
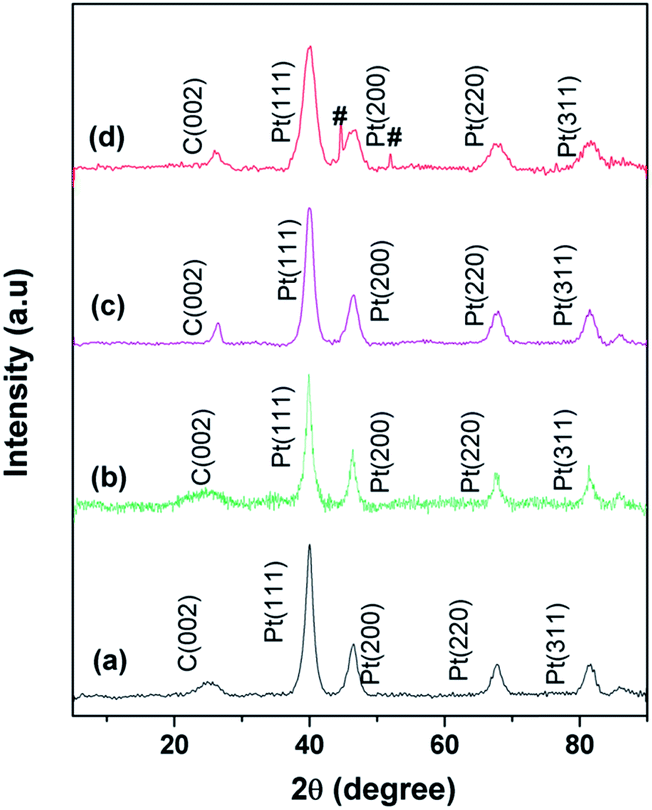
Fig. 3 shows the XRD patterns of (a) graphite, (b) the as-prepared GO, (c) functionalized GO (f-GO), (d) reduced f-GO (Rf-GO), (e) MWNTs and (f) the hybrid structure GCS. Characteristic (002) graphitic peak shifting from 26° to 11° (Fig. 3a and b) confirms the formation of GO from graphite with an enhancement in d spacing from graphitic 0.34 nm to 0.83 nm for the GO sample. A backward shift in the (002) peak can be seen for f-GO (Fig. 3c), revealing the chemical modification of GO by acid treatment and that subsequent polymer incorporation increases the d spacing between the carbon layers to 0.94 nm. An additional peak (marked as *) around 19° for f-GO is attributed to the presence of PEG on the surfaces of GO resulting from surface modification.40 Fig. 3d shows the diffraction pattern of reduced f-GO (Rf-GO); the decrease in the interlayer spacing to 0.37 nm suggests the removal of oxygen-containing functional groups, and the broadened peak depicts the amorphous nature of Rf-GO. Fig. 3e shows the XRD pattern for MWNTs; the sharp (002) peak shows the presence of highly crystalline graphitic structure in MWNTs. The absence of a peak at 11° for Fig. 3d and f proves the complete reduction of f-GO to graphene sheets, and the peak marked ‘#’ in Fig. 3f is due to the presence of a small amount of unused MmNi3 hydride catalyst as impurity. In Fig. 3f, the sharp peak at (002) originates from the presence of MWNTs, and the broad peak at (002) corresponds to the amorphous graphene of hybrid structure.
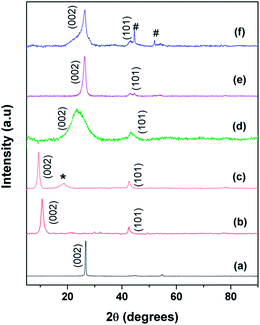 |
| | Fig. 3 XRD spectra of (a) graphite, (b) GO, (c) f-GO, (d) Rf-GO, (e) MWNTs, (f) GCS. | |
XRD patterns for commercial Pt/C, Pt/Rf-GO, Pt/MWNT and Pt/GCS are shown in Fig. 4. The characteristic graphite peak (002) at 26° in these composites confirms the presence of carbon. The (111), (200), (220) and (311) XRD peaks at around ∼40°, 46°, 67° and 81°, respectively, in all Pt incorporated catalyst supports confirm the presence of Pt in the face-centered cubic crystal structure. The broadened Pt peaks show that particles are nanodimensional; crystallite sizes were calculated using the following Scherer equation44
| |
 | (1) |
where
L is the crystallite size of the sample;
λ is the wavelength of the X-ray used;
B is the diffraction peak width at half maxima in radians; and
θ is the value at maximum peak position (see calculated values in
Table 2).
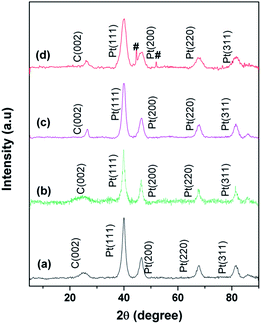 |
| | Fig. 4 XRD patterns for (a) commercial Pt/C, (b) Pt/Rf-GO, (c) Pt/MWNT, (d) Pt/GCS. | |
Table 2 Comparison of crystallite size (XRD), particle size (TEM) and electrochemical surface area (ECSA) of the employed electrocatalyst
| Electrocatalyst |
Avg. crystallite size (L) from XRD (nm) |
Avg. particle size from TEM (nm) |
ECSA (m2 g−1) |
| Pt/C |
6.6 |
5.4 |
25.1 |
| Pt/MWNT |
6.4 |
6.5 |
38.6 |
| Pt/Rf-GO |
7.5 |
5.9 |
37.0 |
| Pt/GCS |
3.6 |
3.5 |
50.7 |
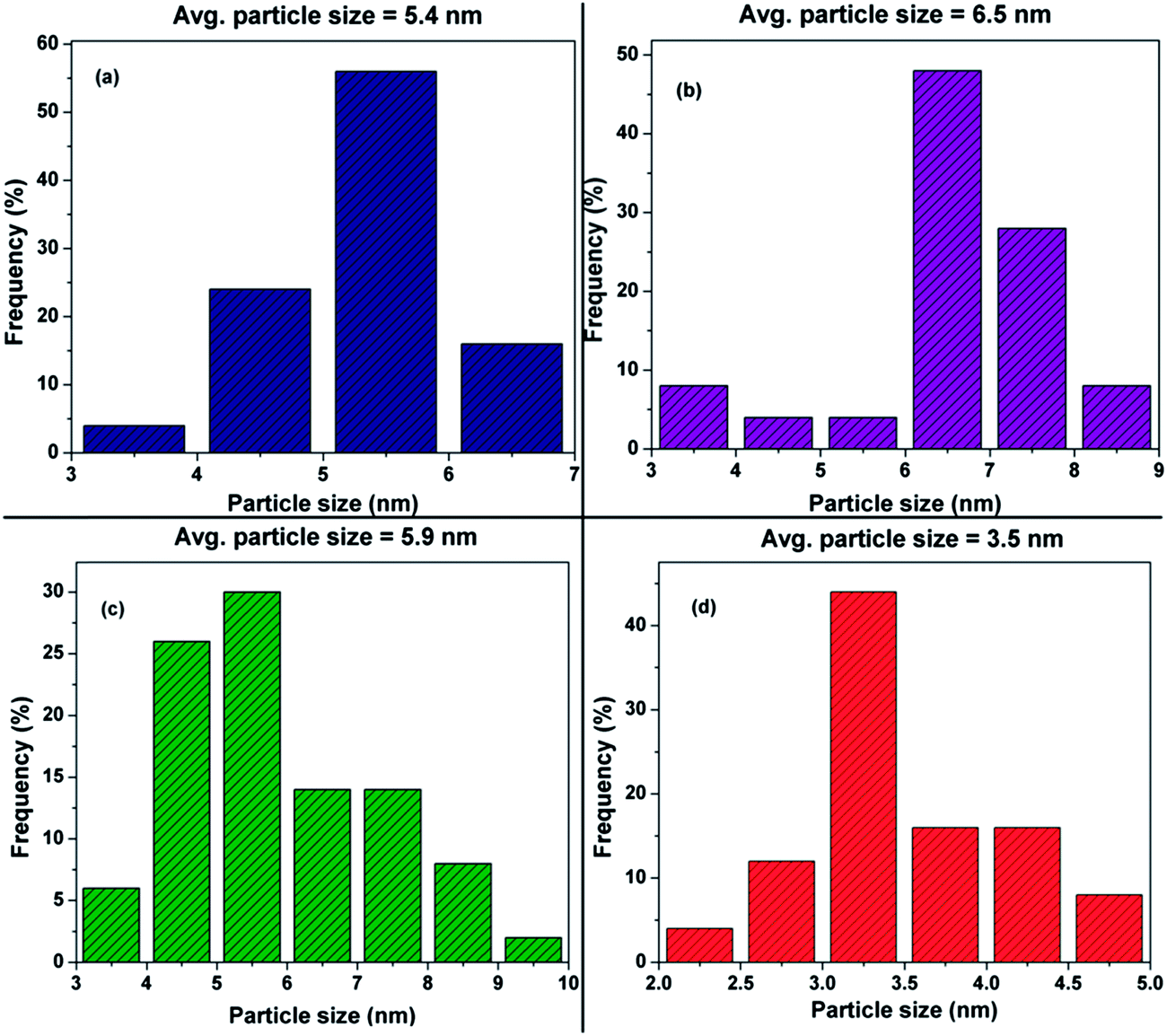
Fig. 5 shows the TEM images of commercial Pt/C, synthesized Pt/MWNT and Pt/Rf-GO; these images exhibit the uniform dispersion of Pt nanoparticles over the support materials The diameter of the synthesized MWNTs is around 40–60 nm. The layered graphene sheets can be seen in Fig. 5c. TEM images of the synthesized Pt/GCS are shown in Fig. 6. Fig. 6a–c show the sandwich-like homogeneous distribution of MWNTs and Rf-GO, with uniform Pt dispersion on the support material and without any agglomeration. The surface modification of GO (f-GO) lowers the speed of removal of the functional groups as compared with pristine GO at reduction, resulting in less-wrinkled surfaces (Fig. 5c and 6); Fig. 6c shows the high magnification image of the boxed portion in Fig. 6b. The particle size estimated from the TEM image is comparable with that of obtained by XRD (Table 2). The dispersion of Pt on the GCS hybrid structure yields the smallest particle size (∼3.5 nm) as compared to other supports such as Vulcan carbon, graphene and MWNTs. Particle size distributions for different Pt supports are shown by the histograms in Fig. 7.
 |
| | Fig. 5 TEM image of (a) commercial Pt/C, (b) Pt/MWNT and (c) Pt/Rf-GO. | |
 |
| | Fig. 6 (a–c) TEM and (d) HRTEM image of Pt/GCS. | |
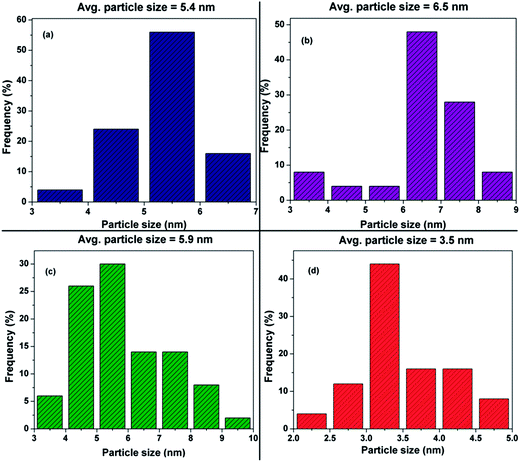 |
| | Fig. 7 Histograms for (a) commercial Pt/C, (b) Pt/MWNT, (c) Pt/Rf-GO and (d) Pt/GCS. | |
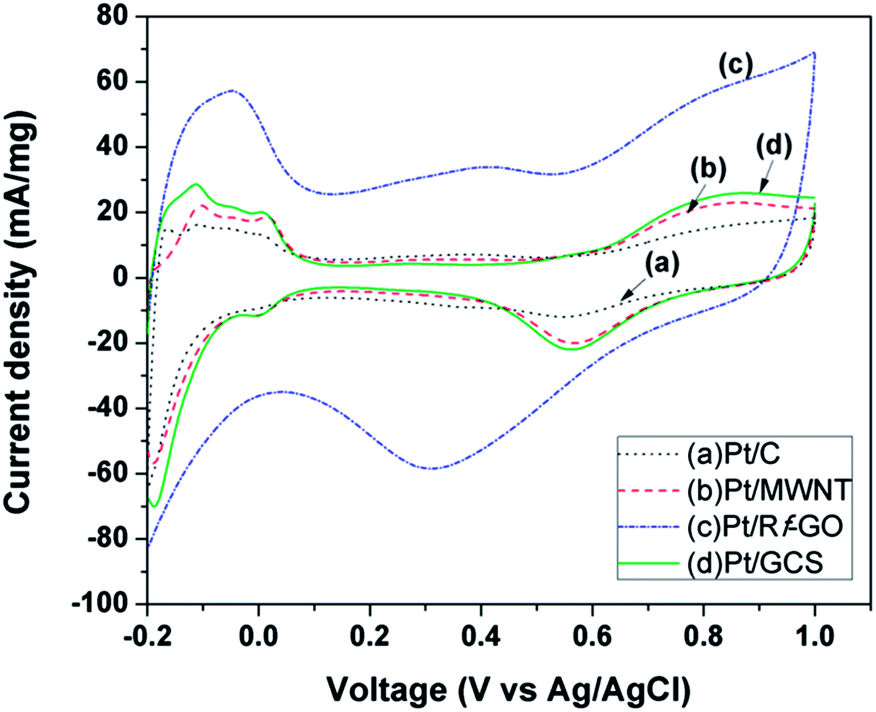
Fig. 8 shows the cyclic voltammograms for commercial Pt/C (20%, Arora Matthey LTD, India) synthesized Pt/Rf-GO, Pt/MWNT and Pt/GCS in the voltage range −0.2 V to 1 V at a scan rate of 50 mV s−1. Measurements were carried out in 1 M H2SO4 solution. Peaks in the region 0.2 V to 0.1 V represents the atomic hydrogen adsorption–desorption on the Pt surface, giving the electrochemical surface area (ECSA) of catalyst45 using the equation
where
Qh is the measured active catalyst surface area, calculated as the mean value of charge exchanges during electro-adsorption and desorption of H
2 on the Pt sites;
45 and
Qm is the adsorption charge for atomically smooth surface area,
i.e., the charge required to oxidize a monolayer of H
2 on the Pt site;
46 both are measured in μC cm
−2. For Pt, the value of
Qm is 210 μC cm
−2.
45 E.L represents the electrode loading in g
Pt cm
−2, which was controlled for each of the catalysts to give the ECSA in m
2 g
Pt−1. The calculated ECSA values after correcting for the electric double layer contribution (formed between electrode and electrolyte) are 25.1, 38.6, 37 and 50.7 m
2 g
Pt−1 for Pt/C, Pt/MWNT, Pt/Rf-GO and Pt/GCS, respectively. In
Fig. 8, formation and abolition of platinum oxide is clearly visible in the potential range 0.4 V to 1 V. The reduction peak potential for Pt-OH
ad as seen from
Fig. 8 is forward shifted for Pt/GCS as compared to Pt/C, suggesting weakened chemical adsorption for oxygen-containing species in the Pt/GCS hybrid catalyst, giving large number of adsorption sites for ORR. The high ECSA of the synthesized Pt/GCS can be due to the homogeneous distribution of high surface area 2-D graphene and high conductive MWNTs in the hybrid support material that lead to the uniform dispersion of Pt nanoparticles, fine binding of Pt with the support, and reduced oxophilicity. The high ECSA of Pt/GCS signifies the availability of a greater number of Pt sites for hydrogen adsorption–desorption, leading to high electrochemical performance.
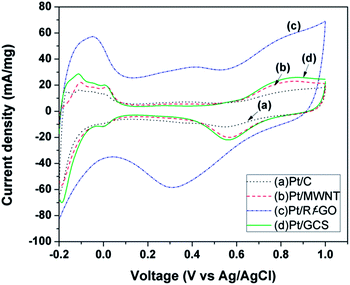 |
| | Fig. 8 Cyclic voltammogram of (a) Pt/C, (b) Pt/MWNT, (c) Pt/Rf-GO, and (d) Pt/GCS in 1 M H2SO4 at 50 mV s−1 scan rate. | |
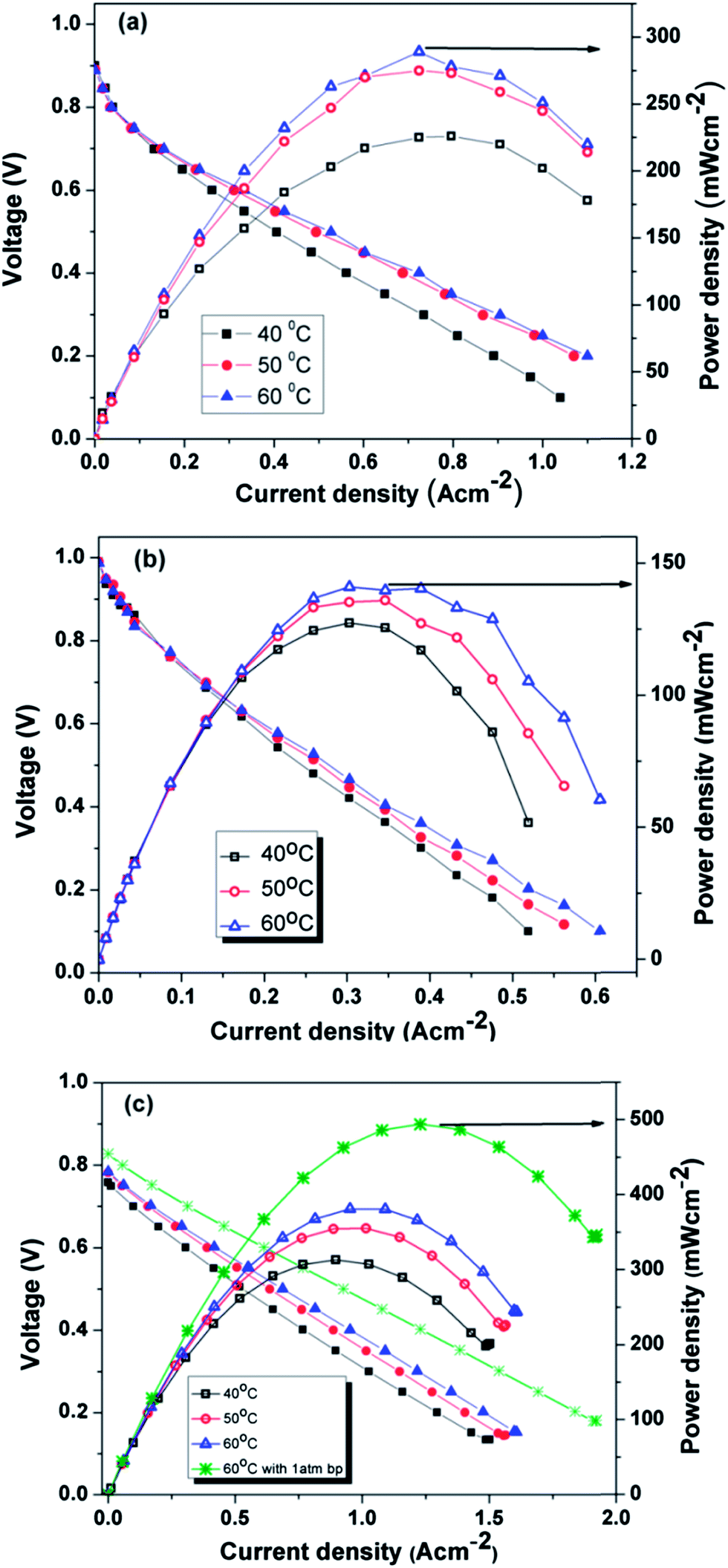
Fig. 9a–c show the polarization curves at three different temperatures (40 °C, 50 °C and 60 °C) for MEA 1(Pt/C), MEA 2 (Pt/Rf-GO) and MEA 3 (Pt/GCS), respectively, with Pt/MWNT as the anode electrocatalyst. For the polarization studies, the cells were first activated between open circuit potential (OCP) and 0.1 V to trigger the catalyst for ORR. In the polarization curve, there is a decrement in cell voltage with an increase in current density, which is attributable to the three known polarization losses: activation, ohmic and mass transport losses. At low current densities, the amount of energy needed to initiate the electrochemical reactions (ORR and HOR) in both the electrodes is used at the expense of cell voltage and known as activation loss. Ohmic loss, the typical linear portion of the plot, is attributed to the resistance coming from all the fuel cell assembly parts such as current collector plates, electrodes and electrolyte; it is the performance-limiting step for PEMFC. In the high current density regime, the depletion of gases at the concerned electrodes and the resulting drop in partial pressure at the reactant site leads to the mass transport or concentration loss. A fuel cell can perform better by reducing all these losses (Table 3).
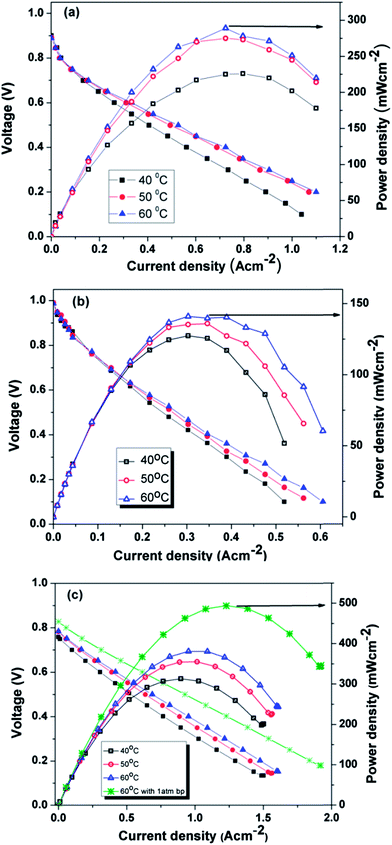 |
| | Fig. 9 Polarization curves for (a) Pt/C, (b) Pt/Rf-GO and (c) Pt/GCS as cathode catalysts with Pt/MWNT as anode electrocatalyst. | |
Table 3 Fuel cell performance of different cathode catalysts with Pt/MWNT as the anode catalyst at 60 °C
| Cathode material |
Current density at 0.6 V (mA cm−2) |
OCP (V) |
Max power density (mW cm−2) |
| Pt/C |
276 |
0.889 |
289 |
| Pt/Rf-GO |
200 |
0.987 |
141 |
| Pt/MWNT47 |
428 (0.54 V) |
0.981 |
— |
| Pt/GCS |
613 |
0.827 |
495 |
Cathode catalysts such as commercial Pt/C, synthesized Pt/Rf-GO and Pt/GCS showed a current density of 276, 200 and 430 mA cm−2 at 0.6 V and 60 °C. The maximum power densities achieved were 289 mW cm−2, 141 mW cm−2 and 382 mW cm−2 at 60 °C without any back pressure for Pt/C, Pt/Rf-GO and Pt/GCS, respectively. The Pt/C performances in this study are less than the values reported by Gasteiger et al.;6 this is mainly due to large discrepancies in the operating conditions for PEMFC testing. The present study has been conducted at a low temperature (60 °C) and without back pressure, whereas the reported one by Gasteiger et al. was carried out at 80 °C and 150 kPa back pressure with different fuel inlet pressures. In the present study, Pt/GCS shows a maximum of 495 mW cm−2 at 60 °C with 1 atm backpressure. A higher current density of 580 mA cm−2 was achieved for Pt/GCS as compared to 428 mA cm−2 at 540 mV for a reported Pt/MWNT at 60 °C.47 The reported Pt/MWNT as the cathode catalyst showed 0.64 V at 300 mA cm−2 with 67 mV dec−1 Tafel slope. MWNTs have already been reported to be more durable compared to Vulcan carbon.48 The high electrical conductivity and tubular morphology of these MWNTs make it a good support material.49 A recent study on Pt supported on PDDA polymer-functionalized graphene50 showed higher performance compared to Pt on pristine graphene31 (maximum power density, 120 mW cm−2). Compared to the reported Pt-PDDA Graphene PEMFC performance,31 the Pt/GCS hybrid catalyst shows excellent performance. The above discussion shows that this combination of Rf-GO and MWNTs provides an impressive PEMFC performance compared with commercial Pt/C, Pt/MWNT and Pt/Rf-GO.
To analyze further, the kinetic parameters were calculated by fitting the polarization plot to the semi-empirical formula51
| | |
Ecell = Eo − blog(I) − RI
| (3) |
where
Ecell and
I are the measured cell potential and current, respectively.
Eo is a constant that depends on the catalysts used and cell operating conditions;
b is the Tafel slope and
R is the total dc resistance.
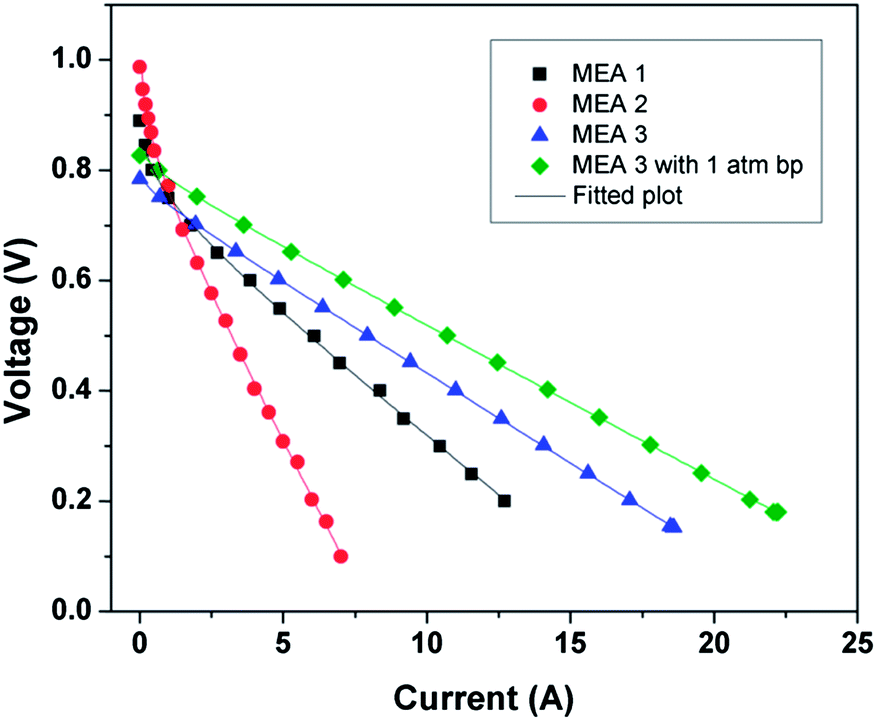
Fig. 10 shows the theoretical fitted plot for the polarization data. The Tafel slope [
d(
Ecell)/
d(log(
I))] was calculated from low current density region; the values are listed in
Table 4. The lowest Tafel slope for the Pt/GCS hybrid electrocatalyst (67.4 mV dec
−1) indicates that ORR is mainly taking place by 4-electron transfer, resulting an enhanced performance as compared to the other catalysts. Also, the dc resistance value for Pt/GCS is very small at 0.027 Ω. Thus, this small resistance value and low Tafel slope support the high performance of Pt/GCS hybrid electrocatalyst.
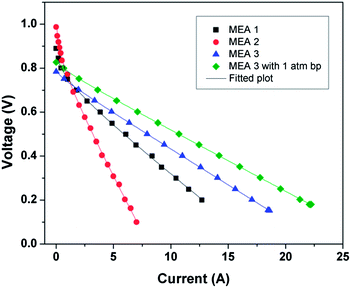 |
| | Fig. 10 Theoretical fit for polarization plots for all MEAs at 60 °C. | |
Table 4 Kinetic parameters for fuel cells obtained by fitting the polarization plots to the semi-empirical formula: Ecell = Eo − blog(I) − RI
| Cathode electrocatalyst |
Kinetic parameters from semi empirical relation, Ecell = Eo − blog(I) − RI |
| b (mV dec−1) |
R (Ω) |
| Pt/C |
72.2 |
0.04 |
| Pt/Rf-GO |
93.4 |
0.094 |
| Pt/GCS |
67.4 |
0.027 |
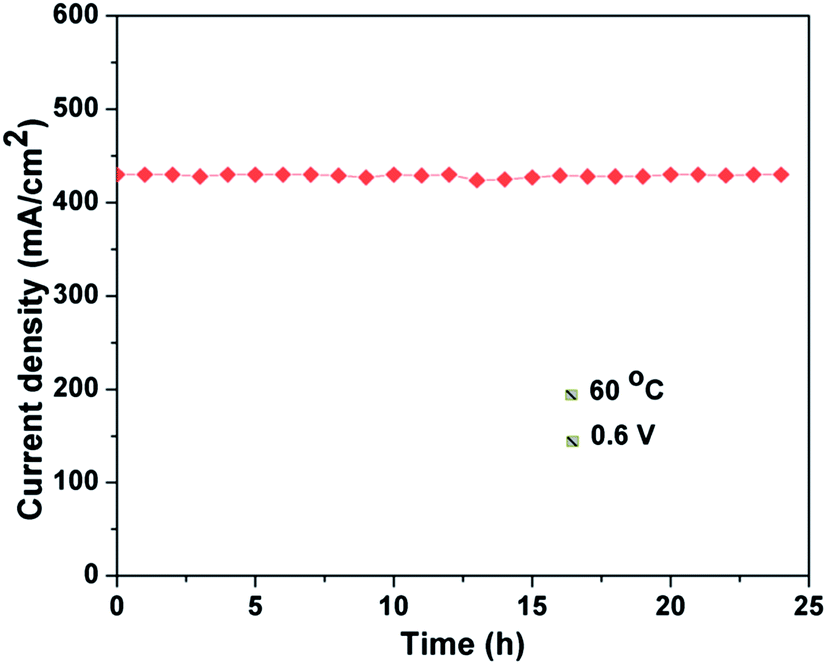
Fig. 11 shows the stability test for the Pt/GCS hybrid cathode catalyst with Pt/MWNT as anode catalyst. The current density was recorded while keeping the voltage fixed at 0.6 V at 60 °C. The cell showed constant current with durable performance for a test run of 24 h. In brief, the uniform dispersion of optimal-sized Pt nanoparticles (3–4 nm) on the conducting, high surface area support materials with strong binding provides the enhanced ORR activity and high durability in PEMFC. The Pt/GCS cathode catalyst performed 71% more effectively than the commercial Pt/C catalyst.
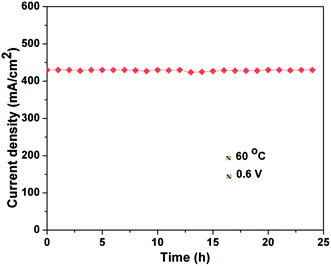 |
| | Fig. 11 Stability test for Pt/GCS cathode catalyst with Pt/MWNT as anode catalyst at 60 °C and 0.6 V. | |
Conclusions
The present study demonstrates a novel synthesis for a chemically modified few-layered and less-wrinkled reduced graphene oxide–MWNT hybrid structure as a cathode catalyst support for PEMFC. In situ growth of the support material from surface-passivized GO and a MmNi3 hydride catalyst produced a homogeneous distribution of 1D MWNT and 2D graphene sheets, giving a high surface area platform for uniform Pt nanoparticle dispersion. Electrochemical measurements show that Pt/GCS has a high electrochemical surface area and excellent ORR activity than commercial Pt/C, Pt/MWNT and Pt/Rf-GO electrocatalysts. The Pt/GCS cathode electrocatalyst gives a maximum PEMFC performance of ∼500 mW cm−2 at 60 °C, 71% higher than that of the commercial Pt/C catalyst.
Acknowledgements
The authors acknowledge the support from DST, Government of India and IITM, Chennai, India.
References
- S. Mekhilef, R. Saidur and A. Safari, Renewable Sustainable Energy Rev., 2012, 16, 981–989 CrossRef CAS PubMed.
- L. Zhang, J. Zhang, D. P. Wilkinson and H. Wang, J. Power Sources, 2006, 156, 171–182 CrossRef CAS PubMed.
- K. Sopian and W. R. Wan Daud, Renewable Energy, 2006, 31, 719–727 CrossRef CAS PubMed.
- S. J. Peighambardoust, S. Rowshanzamir and M. Amjadi, Int. J. Hydrogen Energy, 2010, 35, 9349–9384 CrossRef CAS PubMed.
- X. Yuan, X. Zeng, H.-J. Zhang, Z.-F. Ma and C.-Y. Wang, J. Am. Chem. Soc., 2010, 132, 1754–1755 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS PubMed.
- V. Mazumder, Y. Lee and S. Sun, Adv. Funct. Mater., 2010, 20, 1224–1231 CrossRef CAS.
- F. J. Nores-Pondal, I. M. J. Vilella, H. Troiani, M. Granada, S. R. de Miguel, O. A. Scelza and H. R. Corti, Int. J. Hydrogen Energy, 2009, 34, 8193–8203 CrossRef CAS PubMed.
- X. Cheng, B. Yi, M. Han, J. Zhang, Y. Qiao and J. Yu, J. Power Sources, 1999, 79, 75–81 CrossRef CAS.
- S. Mukerjee, S. Srinivasan, M. P. Soriaga and J. McBreen, J. Electrochem. Soc., 1995, 142, 1409–1422 CrossRef CAS PubMed.
- C. Venkateswara Rao and B. Viswanathan, J. Phys. Chem. C, 2009, 113, 18907–18913 CAS.
- V. DiNoto, E. Negro, R. Gliubizzi, S. Lavina, G. Pace, S. Gross and C. Maccato, Adv. Funct. Mater., 2007, 17, 3626–3638 CrossRef.
- M. A. García-Contreras, S. M. Fernández-Valverde, J. R. Vargas-García, M. A. Cortés-Jácome, J. A. Toledo-Antonio and C. Ángeles-Chavez, Int. J. Hydrogen Energy, 2008, 33, 6672–6680 CrossRef PubMed.
- Y.-H. Cho, T.-Y. Jeon, J. W. Lim, Y.-H. Cho, M. Ahnb, N. Jung, S. J. Yoo, W.-S. Yoon and Y.-E. Sung, Int. J. Hydrogen Energy, 2011, 36, 4394–4399 CrossRef CAS PubMed.
- C. S. Rao, D. M. Singh, R. Sekhar and J. Rangarajan, Int. J. Hydrogen Energy, 2011, 36, 14805–14814 CrossRef CAS PubMed.
- F. Kadirgan, A. M. Kannan, T. Atilan, S. Beyhan, S. S. Ozenler, S. Suzer and A. Yörür, Int. J. Hydrogen Energy, 2009, 34, 9450–9460 CrossRef CAS PubMed.
- E. B. Fox and H. R. Colon-Mercado, Int. J. Hydrogen Energy, 2010, 35, 3280–3286 CrossRef CAS PubMed.
- A. J. Plomp, D. S. Su, K. P. d. Jong and J. H. Bitter, J. Phys. Chem. C, 2009, 113, 9865–9869 CAS.
- I. Kvande, J. Zhu, T.-J. Zhao, N. Hammer, M. Rønning, S. Raaen, J. C. Walmsley and D. Chen, J. Phys. Chem. C, 2010, 114, 1752–1762 CAS.
- S.-H. Liu, C.-C. Chiang, M.-T. Wu and S.-B. Liu, Int. J. Hydrogen Energy, 2010, 35, 8149–8154 CrossRef CAS PubMed.
- R. I. Jafri and S. Ramaprabhu, Int. J. Hydrogen Energy, 2010, 35, 1339–1346 CrossRef CAS PubMed.
- C. Arbizzani, S. Righi, F. Soavi and M. Mastragostino, Int. J. Hydrogen Energy, 2011, 36, 5038–5046 CrossRef CAS PubMed.
- N. Jha, R. I. Jafri, N. Rajalakshmi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2011, 36, 7284–7290 CrossRef CAS PubMed.
- D. Sebastián, J. C. Calderón, J. A. González-Expósito, E. Pastor, M. V. Martínez-Huerta, I. Suelves, R. Moliner and M. J. Lázaro, Int. J. Hydrogen Energy, 2010, 35, 9934–9942 CrossRef PubMed.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 CAS.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990–7995 CAS.
- Y. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792–6797 CrossRef CAS.
- S. S. Jyothirmayee Aravind, R. Imran Jafri, N. Rajalakshmi and S. Ramaprabhu, J. Mater. Chem., 2011, 21, 18199–18204 RSC.
- R. I. Jafri, T. Arockiados, N. Rajalakshmi and S. Ramaprabhu, J. Electrochem. Soc., 2010, 157, B874–B879 CrossRef CAS PubMed.
- B. P. Vinayan, R. Nagar, V. Raman, N. Rajalakshmi, K. S. Dhathathreyan and S. Ramaprabhu, J. Mater. Chem., 2012, 22, 9949–9956 RSC.
- S. S. J. Aravind and S. Ramaprabhu, ACS Appl. Mater. Interfaces, 2012, 4, 3805–3810 CAS.
- B. P. Vinayan, R. I. Jafri, R. Nagar, N. Rajalakshmi, K. Sethupathi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2012, 37, 412–421 CrossRef CAS PubMed.
- J. Qi, L. Jiang, M. Jing, Q. Tang and G. Sun, Int. J. Hydrogen Energy, 2011, 36, 10490–10501 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. P. Vinayan, R. Nagar, K. Sethupathi and S. Ramaprabhu, J. Phys. Chem. C, 2011, 115, 15679–15685 Search PubMed.
- A. L. M. Reddy, M. M. Shaijumon and S. Ramaprabhu, Nanotechnology, 2006, 17, 5299 CrossRef CAS.
- J.-C. Charlier, A. De Vita, X. Blase and R. Car, Science, 1997, 275, 647–649 CrossRef.
- Y. Saito, T. Yoshikawa, M. Inagaki, M. Tomita and T. Hayashi, Chem. Phys. Lett., 1993, 204, 277–282 CrossRef CAS.
- R. S. Wagner and W. C. Ellis, Appl. Phys. Lett., 1964, 4, 89–90 CrossRef CAS PubMed.
- C. Wang, L. Feng, H. Yang, G. Xin, W. Li, J. Zheng, W. Tian and X. Li, Phys. Chem. Chem. Phys., 2012, 14, 13233–13238 RSC.
- P. M. Ajayan, Chem. Rev., 1999, 99, 1787–1800 CrossRef CAS PubMed.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS PubMed.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- A. Pozio, M. De Francesco, A. Cemmi, F. Cardellini and L. Giorgi, J. Power Sources, 2002, 105, 13–19 CrossRef CAS.
- S. J. Lee, S. Mukerjee, J. McBreen, Y. W. Rho, Y. T. Kho and T. H. Lee, Electrochim. Acta, 1998, 43, 3693–3701 CrossRef CAS.
- A. L. M. Reddy, N. Rajalakshmi, S. Ramaprabhu and M. M. Shaijumon, J. Fuel Cell Sci. Technol., 2009, 7, 021001 CrossRef.
- X. Wang, W. Li, Z. Chen, M. Waje and Y. Yan, J. Power Sources, 2006, 158, 154–159 CrossRef CAS PubMed.
- W. Li, X. Wang, Z. Chen, M. Waje and Y. Yan, Langmuir, 2005, 21, 9386–9389 CrossRef CAS PubMed.
- J. Y. Park and S. Kim, Int. J. Hydrogen Energy, 2013, 38, 6275–6282 CrossRef CAS PubMed.
- S. Srinivasan, E. A. Ticianelli, C. R. Derouin and A. Redondo, J. Power Sources, 1988, 22, 359–375 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.