
Effect of branching in remote substituents on light emission and stability of chemiluminescent acridinium esters†
Abstract
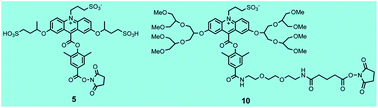
Acridinium dimethylphenyl esters are widely used as chemiluminescent labels in automated immunoassays for clinical diagnostics in Siemens Healthcare Diagnostics' ADVIA Centaur® systems. Light emission from these labels and their conjugates is triggered with alkaline peroxide. Excited state acridone is believed to be the light emitting species that is formed from the initial peroxide adduct which subsequently undergoes a series of reactions leading to scission of the phenolic ester bond. Dioxetane and/or dioxetanone intermediates have been proposed as immediate precursors of excited state acridone. Despite the fact that acridinium esters have been widely used as chemiluminescent labels for decades, a substantive theoretical framework to guide acridinium ester design with improved properties over the basic structure is unavailable. We have relied on a more empirical approach to devise new acridinium esters with improved stability, higher light yield, fast light emission, low non-specific binding and improved immunoassay performance. In the current study, we have investigated the effect of branching in remote alkoxy substituents attached to C-2 and C-7 of the acridinium ring on light emission and chemiluminescence stability of two acridinium esters. We selected two, high light output acridinium dimethylphenyl esters that we described previously as a basis for these studies and report the synthesis of two new C-2 and C-7 alkoxy-substituted labels, compounds 5 and 10, with improved chemiluminescence stability and faster light emission respectively. Compound 5 exhibited better long term stability at both pH 6 and 7.4 (≥10%) compared to its unbranched counterpart compound 11 whereas compound 10 with branched hexa(ethylene) glycol substituents exhibited >4-fold faster light emission compared to its unbranched counterpart 12. Both parameters are important for immunoassay performance in automated instruments.
 Please wait while we load your content...
Please wait while we load your content...

