DOI:
10.1039/C4RA02433H
(Paper)
RSC Adv., 2014,
4, 21830-21839
Synthesis and self-assembly of water-soluble polythiophene-graft-poly(ethylene oxide) copolymers
Received
20th March 2014
, Accepted 30th April 2014
First published on 7th May 2014
Abstract
In this study, we synthesized amphiphilic poly(3-hexylthiophene)-graft-poly(ethylene oxide) (P3HT-g-PEO) rod–coil conjugated random copolymers through oxidative polymerization with FeCl3 and facile click chemistry and characterized them using 1H nuclear magnetic resonance spectroscopy, size exclusion chromatography, differential scanning calorimetry (DSC), X-ray photoelectron spectroscopy, UV-Vis spectroscopy, and fluorescence spectroscopy. We then used atomic force microscopy, transmission electron microscopy, and dynamic light scattering to investigate the self-assembled structures formed from these amphiphilic random copolymers in solution and in the bulk state. In the bulk state, DSC analyses revealed that after PEO had been grafted onto P3HT, the crystallization temperature of PEO decreased from +20 to −26 °C as a result of hard confinement of microphase separation in the copolymer system. In addition, we found that the amphiphilic conjugated random copolymers could form micelle structures in the DMF–water system.
Introduction
Conjugated polymers have received a great deal of attention in recent years as alternatives to inorganic single-crystalline semiconductors because of their similar properties and solution processability.1–3 Rod–coil block semiconducting polymers are particularly interesting because they combine the optical and electronic properties of conjugated polymers with the fascinating self-assembly behavior of block copolymers.4 In rod–coil block copolymers, flexible coil-like chains are covalently bonded to rod-like chains to tailor the structures of the conjugated blocks; their self-assembly relies on four thermodynamic parameters: the Flory–Huggins strength of segregation(XN) where N is the molecular length which parameterizes the interactions between chemically dissimilar blocks; the Maier–Saupe interaction relating the rod–rod alignment tendency (μN); the volume fraction of coil and the geometrical asymmetry of the system.5 These parameters can give rise to unconventional phase-separated morphologies, which remain relatively unexplored and less understood than those obtained from classical coil–coil block copolymers. Increasingly, solvent-induced ordering is being used to tailor the nano-domain morphologies in block copolymers.6,7 For a given copolymer system, a particular solvent may be classified as neutral or selective, according to whether it is a good solvent for both blocks (neutral), or a good solvent for one but a poor or non solvent for the others (selective).8 In general, a neutral solvent distributes itself nearly equally between micro domains and can screen unfavorable contacts between different blocks. Among conjugated polymers, polythiophene (PT) is one of the most promising because of its ability to conduct electrons and its controllable electrochemical behavior. For some time, its applicability was limited by its insolubility in many organic solvents, due to its highly π-conjugated structure. To solve this problem, alkyl chains can be added at the C3 positions of the thiophene units. The resulting poly(3-alkylthiophene)s (P3ATS) are processable conducting polymers possessing modifiable electronic properties; they can be fully characterized through chemical and physical means.9 P3ATS exhibiting stability in the doped state can be achieved by releasing the side alkyl chains crowding along the backbone or by introducing alkoxy groups. A monomer containing two methylene groups between the thiophene ring and the first oxygen atom, allow to obtain highly stable polymers.10,11 Regioregular P3ATS are among the most promising conjugated polymers because of their good solubility, chemical stability, excellent electronic properties, and ease of preparation.12 These characteristics make them readily accessible for optoelectronic device applications, such as organic field-effect transistors,13 photovoltaic cells,14,15 and sensors.16 Several P3AT-based rod–coil block copolymers have been reported recently, including poly(3-hexylthiophene)-block-poly(vinyl pyridine) (P3HT-b-PVP),17 poly(3-hexylthiophene)-block-poly(methyl methacrylate) (P3HT-b-PMMA),18,19 poly(3-hexylthiophene)-block-poly lactide (P3HT-b-PLA),20,21 poly(3-hexylthiophene)-block-poly(ethylene oxide) (P3HT-b-PEO),22,23 and poly(3-hexylthiophene)-block-polystyrene (P3HT-b-PS).24,25 Such copolymers are usually synthesized using a “grafting from” approach, where an end-functionalized PT is used as a macroinitiator for the polymerization of a second block, or a “grafting to” approach in the blocks are prepared separately and then linked together. “Click” chemistry between azido and alkynyl groups has become very popular in recent years, because of its high efficiency; since 2008, this reaction has been extended to the preparation of block copolymers containing conjugated segments, including P3HT-b-PS,25 poly(3-hexylthiophene)-block-(poly-γ-benzyl-L-glutamate) (P3HT-b-PBLG),26 and poly(3-hexylthiophene)-block-poly(acrylic acid) (P3HT-b-PAA).27,28 Recently, the diblock copolymer poly(3-hexylthiophene)-block-poly(ethylene oxide) (P3HT-b-PEO) was prepared from the monomer 2,5-dibromohexylthiophene and the use of a click reaction; it can form hierarchical assembled structures of isolated, bundled, and branched nanofibers in solution29 and exhibit simultaneous ionic and electronic conductivity when used in a battery cathode.30
In this study, we report the synthesis and characterization of amphiphilic random PTs grafted to poly (ethylene oxide) (Mn = 1000 g mol−1). We synthesized amphiphilic random PTs using a combination of oxidative polymerization (mediated by FeCl3) and click reactions between azido-grafted random P3HT copolymers and ethynyl-terminated PEO (hydrophilic side chain). First, we prepared two random conjugated copolymers (random P3HT-Br) through FeCl3-mediated oxidative polymerization of various ratios of 3-hexylthiophene and 3-(6-bromohexylthiophene) (Scheme 1) and then treated them with sodium azide (NaN3) to obtain azido-grafted PTs. We then prepared amphiphilic random conjugated copolymers through click reactions of the random P3HT-N3 copolymers with ethynl-terminated PEO (Scheme 1). Herein, we report their characterization, thermal and optical properties, and self-assembled structures.
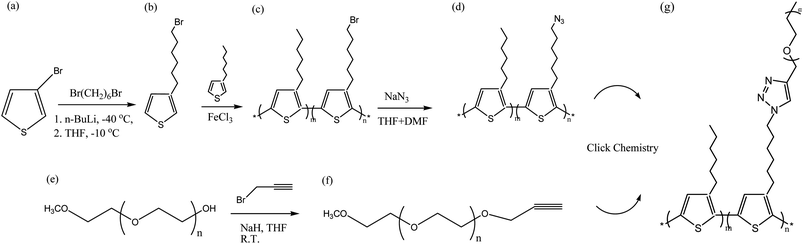 |
| | Scheme 1 Synthesis of P3HT-g-PEO random copolymers through oxidative polymerization and click reactions. | |
Experimental section
Materials
Tetrahydrofuran (THF) and CHCl3 were dried over sodium/benzophenone and distilled under N2 prior to use. Propargyl bromide and 1,6-dibromohexane were purchased from Alfa Aesar. Copper bromide (CuBr) was purified by washing with glacial AcOH overnight, followed by washing with absolute EtOH and Et2O and then drying under vacuum. N,N-Dimethylformamide (DMF), NaN3, pentamethyldiethylenetriamine (PMDETA, 99%), 3-hexylthiophene, 3-bromothiophene, and n-butyl lithium were purchased from Aldrich. 3-(6-Bromohexyl)thiophene31 and propargyl-PEO32 were prepared according to procedures described in the literature.
3-Bromohexylthiophene (1)
A 500 mL three-necked round-bottom flask equipped with a stirrer bar was charged with 3-bromothiophene (30 g, 0.18 mol) and hexane (250 mL). The flask was cooled to −40 °C and the solution was stirred for 10 min. n-BuLi (72 mL, 0.18 mol) was added drop wise via syringe at this temperature. After the mixture had stirred for 10 min, THF (15 mL) was added drop wise via syringe. The solution was stirred for 1 h, the cooling bath was removed, and then the mixture was warmed to −10 °C. 1,6-Dibromohexane (110 mL, 0.72 mol) was added in one portion and then the solution was warmed to room temperature, stirred for 24 h, and then extracted with Et2O (320 mL). The extract was washed with water (3 × 20 mL). The organic phase was dried (anhydrous MgSO4) and concentrated to give a crude product, which was purified through vacuum distillation. Yield: 27 g (59.4%); 1H NMR (500 MHz, CDCl3): δ 7.24 (d, 1H), 6.92 (d, 1H), 3.40 (t, 2H), 2.64 (t, 2H), 1.86–1.89 (m, 2H), 1.65–1.69 (m, 2H), 1.56–1.58 (m, 2H), 1.24–1.27 (m, 2H). 13C NMR (400 MHz, CDCl3): δ 26.45, 28.96, 29.19, 30.30, 30.49, 49.06, 102.34, 120.17, 125.45, 128.40, 142.96, 144.65, 151.15, 164.18.
Random P3HT-Br copolymers (P1, P2)
A solution of anhydrous FeCl3 (1.90 g, 11.7 mmol) in dry CHCl3 (20 mL) was added drop wise to a stirred solution of 3-hexylthiophene (0.400 mL, 2.22 mmol) and 6-(3-bromohexylthiophene) (0.390 mL, 2.05 mmol) in dry CHCl3 (15 mL) and then solution was purged with dry Ar for 10 min. The mixture was degassed through three freeze/evacuation cycles. Subsequently, the mixture was stirred for 12 h at room temperature and then poured into MeOH (200 mL). Rotary evaporation of CHCl3 precipitated a crude polymer, which was filtered off, extracted with refluxing MeOH in a Soxhlet extractor for 48 h, and then dried under vacuum. Yield: 0.56 g; 1H NMR (500 MHz, CDCl3): δ 6.14 (s, 1H), 3.51 (t, 2H), 2.62 (t, 4H), 1.82 (t, 2H), 1.5 (m, 4H), 1.31 (m, 2H), 0.88 (t, 3H).
Random P3HT-N3 copolymers
A solution of random P3HT-Br (0.50 g, 0.012 mmol) in THF (20 mL) and DMF (20 mL) was heated under reflux in a two-neck, 250 mL, round-bottom flask equipped with a stirrer bar. NaN3 (4.5 g, 69 mmol) was added in one portion and then the mixture was stirred overnight under reflux. After cooling, the reaction was quenched through the addition of MeOH and CH2Cl2; the solid polymer was collected and washed several times with MeOH. Yield: 0.3 g (49%); 1H NMR (500 MHz, CDCl3): δ 6.14 (s, 1H), 3.2 (t, 2H), 2.62 (t, 4H), 1.82 (t, 2H), 1.5 (m, 4H), 1.31 (m, 4H), 1.29 (m, 8H), 0.88 (t, 3H).
Propargyl-PEO
Poly(ethylene oxide) monomethyl ether (13.3 g, 13.3 mmol) was dried under vacuum at 100 °C for 2 h. After cooling to 0 °C, dry THF (40 mL) and NaH (0.520 g, 21.7 mmol) were added. The mixture was stirred at room temperature for 12 h and then propargyl bromide (2.74 g, 23.0 mmol). The resulting solution was stirred at room temperature for 4 h and then passed through a short column of silica gel (eluent: THF). The clear solution was concentrated and the residue redissolved in MeOH and a small amount of water. This methanolic solution was extracted with hexane to remove excess propargyl bromide. The MeOH was evaporated; Et2O was added and the mixture dried (MgSO4). After filtration and concentration, the product was obtained as a viscous yellow oil. Yield: 75%. 1H NMR (500 MHz, CDCl3): δ 4.14 (d, 2H), 3.6 (br, OCH2CH2O), 3.3 (s, 3H, OCH3), 2.4 (t, 1H, CH). 13C NMR (400 MHz, CDCl3): δ 79.5, 74.5, 71.8, 71.8–69.0 (OCH2CH2O), 59.0, 58.4. GPC: Mn = 819 g mol−1; Mw = 1042 g mol−1; PDI = 1.27.
P3HT-g-PEO random copolymers
A mixture of propargyl-PEO (0.60 g, 0.58 mmol) and random P3HT-N3 (0.30 g) in DMF (10 mL) and THF (10 mL) was purged with a dry Ar for 10 min. PMDETA (41.7 μL, 0.02 mol) was added via syringe, resulting in the mixture becoming homogeneous; the solution was then degassed through three freeze/thaw evacuation cycles. After addition of CuBr (0.002 g, 0.02 mmol), the color of the solution changed gradually from light blue to light green. The solution was heated at 50 °C with stirring under an Ar atmosphere until the azide peak (2092 cm−1) disappeared completely (8 h) from the FTIR spectrum. After cooling to 25 °C, the solution was subjected to vacuum distillation to remove DMF–THF. The residue was purified through precipitation into MeOH; the solid was filtered off and dried under vacuum. Yield: 0.23 g (49%); 1H NMR (500 MHz, CDCl3): δ 7.42 (s, 1H), 6.14 (s, 1H), 4.11 (s, 2H), 3.54 (m, 4H), 3.3 (s, 3H), 2.62 (t, 4H), 1.59 (m, 4H), (t, 2H), 1.59 (m, 4H), 1.29 (m, 10H), 1.31 (m, 4H), 0.88 (t, 3H). GPC: Mn = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1; PDI = 1.69.
000 g mol−1; PDI = 1.69.
Micelle solution
The P3HT-g-PEO random copolymers were first dissolved in DMF, a common solvent (nonselective solvent) for both the P3HT and PEO blocks; a second solvent (water), a good solvent for PEO blocks, was added slowly to the stirred polymer solution, via syringe pump, at a constant rate (typically 1–5 mL h−1). The solution was stirred for 2 days prior to further characterization.
Characterization
1H NMR spectra were recorded at room temperature using a Bruker AM 500 (500 MHz) spectrometer, with the residual proton resonance of the deuterated solvent acting as the internal standard. Molecular weights and molecular weight distributions were determined through gel permeation chromatography (GPC) using a Waters 510 high-performance liquid chromatography (HPLC) system equipped with a 410 differential refractometer and three Ultrastyragel columns (100, 500, and 103 Å) connected in series, with THF as the eluent (flow rate: 0.4 mL min−1). Differential scanning calorimetry (DSC) was performed using a TA-Q20 instrument operated at a scan rate of 20 °C min−1 over a temperature range from −90 to +200 °C under a N2 atmosphere. X-ray photoelectron spectroscopy (XPS) was performed using a PHI Quantera SXM instrument equipped with a 180 °C hemispherical electron energy analyzer and a monochromatized Al Kα (1486.6 eV) source operated at 15 kV and 4 mA. Atomic force microscopy (AFM) was performed using a Veeco Multimode AFM Nanoscope IV apparatus operated in tapping mode. For TEM studies in solution, a drop of the resulting micelle solution was sprayed onto a Cu TEM grid covered with a Formvar supporting film that had been pre-coated with a thin film of carbon. All samples were left to dry at room temperature for 1 day prior to observation. After 1 min, the excess solvent was blotted away using a strip of filter paper. UV-Vis absorption spectra of polymer solutions were recorded using a Jasco V-560 UV-Vis spectrophotometer over the wavelength range 300–800 nm. Fluorescence spectra of polymer solution were recorded using a Jasco FP-750 spectrofluorometer over the wavelength range 450–700 nm. Dynamic light scattering (DLS) was performed using a Malvern Zetasizer Nano Series to determine the size distribution profiles of polymers in solution (DMF–H2O).
Results and discussion
Synthesis of P3HT-g-PEO random copolymers through sequential oxidative polymerization and click chemistry
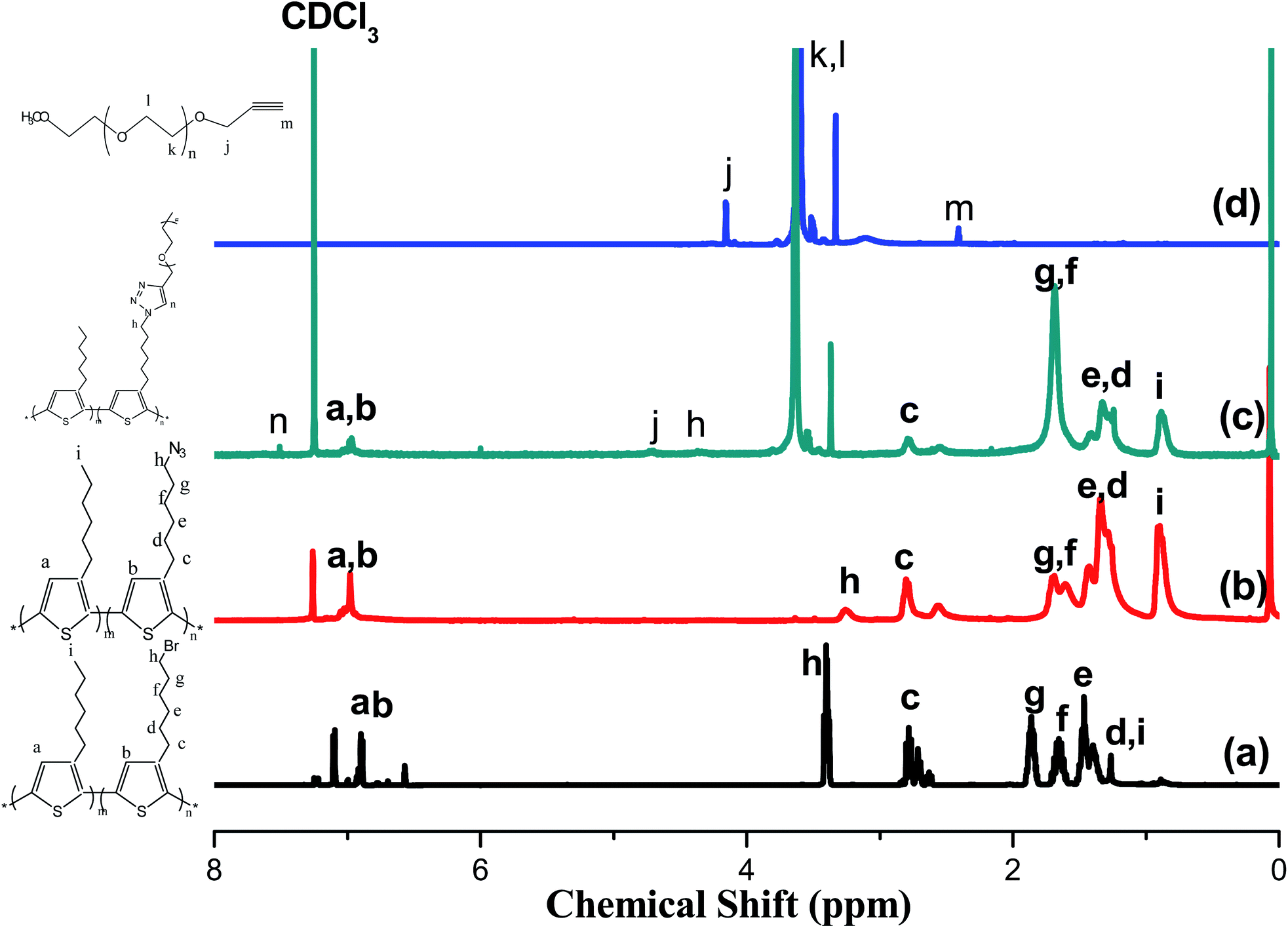
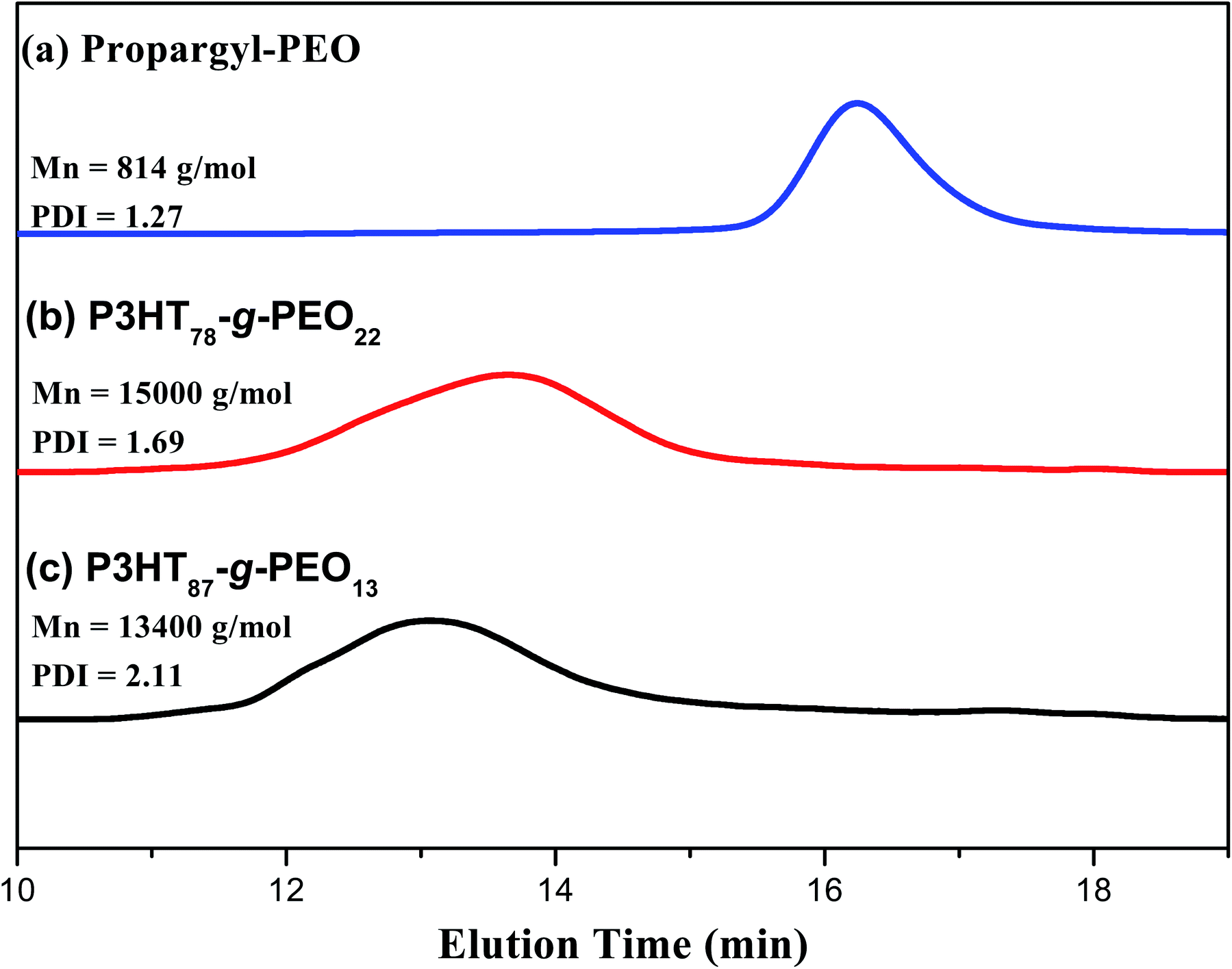
Fig. 1 presents the 1H NMR spectra of P3HT-Br, P3HT-N3, propargyl-PEO, and the P3HT-g-PEO random copolymer. The 1H NMR spectrum of the random P3HT-Br in CDCl3 features [Fig. 1(a)] signals at 6.57–6.84 and 3.43–1.22 ppm corresponding to the aromatic (proton of the thiophene ring) and CH2Br/aliphatic protons, respectively; we assign the singlet at 3.43 ppm to the methylene proton adjacent to the Br group of P3HT-Br. Fig. 1(b) displays the 1H NMR spectrum of random P3HT-N3 in CDCl3; the signal for the CH2 proton adjacent to the azide group of P3HT-N3 appeared as a singlet at 3.24 ppm, shifted upfield from 3.43 ppm of P3HT-Br. The 1H NMR spectrum of propargyl-PEO in CDCl3 displays [Fig. 1(d)] singlets at 4.14, 3.6, 3.3, and 2.4 ppm corresponding to the CH2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, OCH2CH2O, OCH3, and CHC–C protons, respectively. Fig. 1(c) displays the 1H NMR spectrum of the P3HT-g-PEO random copolymer. The singlets at 7.46, 4.3, and 3.6 ppm correspond to the CHC proton on the triazole ring (resulting from the click reaction) and the CH2N and CH2O protons, respectively. Notably, the signal for the CH2N group connected to the azide atoms shifted downfield significantly to 4.30 ppm (from 3.24 ppm for P3HT-N3). Thus, 1H NMR spectroscopy confirmed the successful preparation of the random PTs grafted with PEO units as side chains; the yields of the polymers were, however, not particularly high (<50%). Table 1 summarizes the characteristics of the synthesized random copolymers. GPC analyses (Fig. 2) revealed that the PDIs of the random copolymers were not particularly low after oxidative polymerization. Furthermore, the feed ratios of the two monomers were not the same as those in the random copolymers, as confirmed by 1H NMR spectroscopy. We calculated the repeating monomer units of the P3HT and P3HT-Br segments in the random copolymers through integration of the signals of the methylene protons (Hh) adjacent to the Br atom and the terminal methyl protons (Hi) in the P3HT segment [Fig. 1(a)]. The amphiphilic random PTs exhibited good solubility in common solvents, including CHCl3, THF, toluene, hexane, and water.
C, OCH2CH2O, OCH3, and CHC–C protons, respectively. Fig. 1(c) displays the 1H NMR spectrum of the P3HT-g-PEO random copolymer. The singlets at 7.46, 4.3, and 3.6 ppm correspond to the CHC proton on the triazole ring (resulting from the click reaction) and the CH2N and CH2O protons, respectively. Notably, the signal for the CH2N group connected to the azide atoms shifted downfield significantly to 4.30 ppm (from 3.24 ppm for P3HT-N3). Thus, 1H NMR spectroscopy confirmed the successful preparation of the random PTs grafted with PEO units as side chains; the yields of the polymers were, however, not particularly high (<50%). Table 1 summarizes the characteristics of the synthesized random copolymers. GPC analyses (Fig. 2) revealed that the PDIs of the random copolymers were not particularly low after oxidative polymerization. Furthermore, the feed ratios of the two monomers were not the same as those in the random copolymers, as confirmed by 1H NMR spectroscopy. We calculated the repeating monomer units of the P3HT and P3HT-Br segments in the random copolymers through integration of the signals of the methylene protons (Hh) adjacent to the Br atom and the terminal methyl protons (Hi) in the P3HT segment [Fig. 1(a)]. The amphiphilic random PTs exhibited good solubility in common solvents, including CHCl3, THF, toluene, hexane, and water.
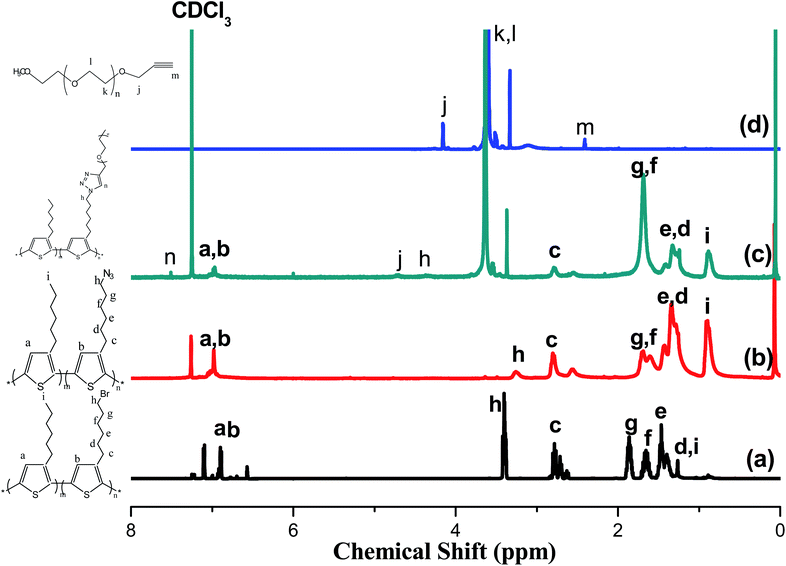 |
| | Fig. 1 1H NMR spectra of (a) P3HT-Br random copolymer, (b) P3HT-N3 random copolymer, (c) P3HT-g-PEO random copolymer, and (d) PEO homopolymer. | |
Table 1 Characteristics of the P3HT and P3HT-g-PEO polymers tested in this study
| Polymer |
Mnb |
Mnb |
PDIb |
| Ratio determined from 1H NMR spectra. Determined through GPC analysis. |
| P3HT |
5082 |
17500 |
3.46 |
| P3HT87-g-PEO13a |
13300 |
28200 |
2.11 |
| P3HT78-g-PEO22a |
15000 |
25455 |
1.69 |
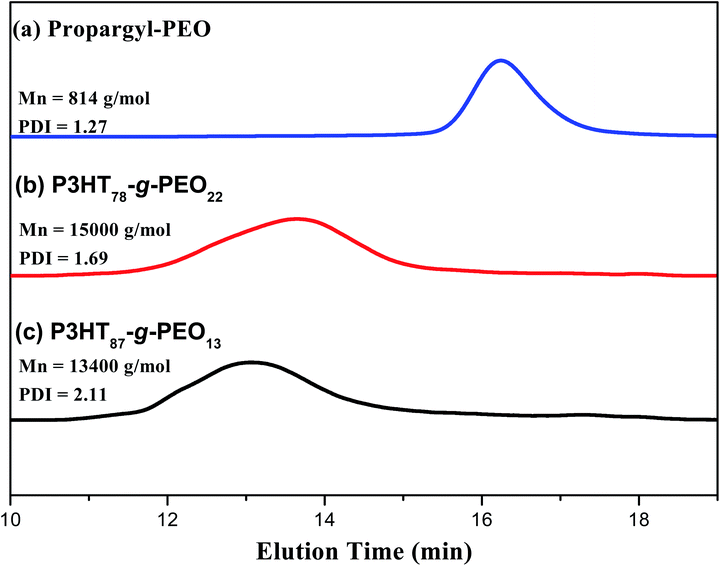 |
| | Fig. 2 GPC traces of (a) PEO homopolymer, (b) P3HT78-g-PEO22 random copolymer, and (c) P3HT87-g-PEO13 random copolymer. | |
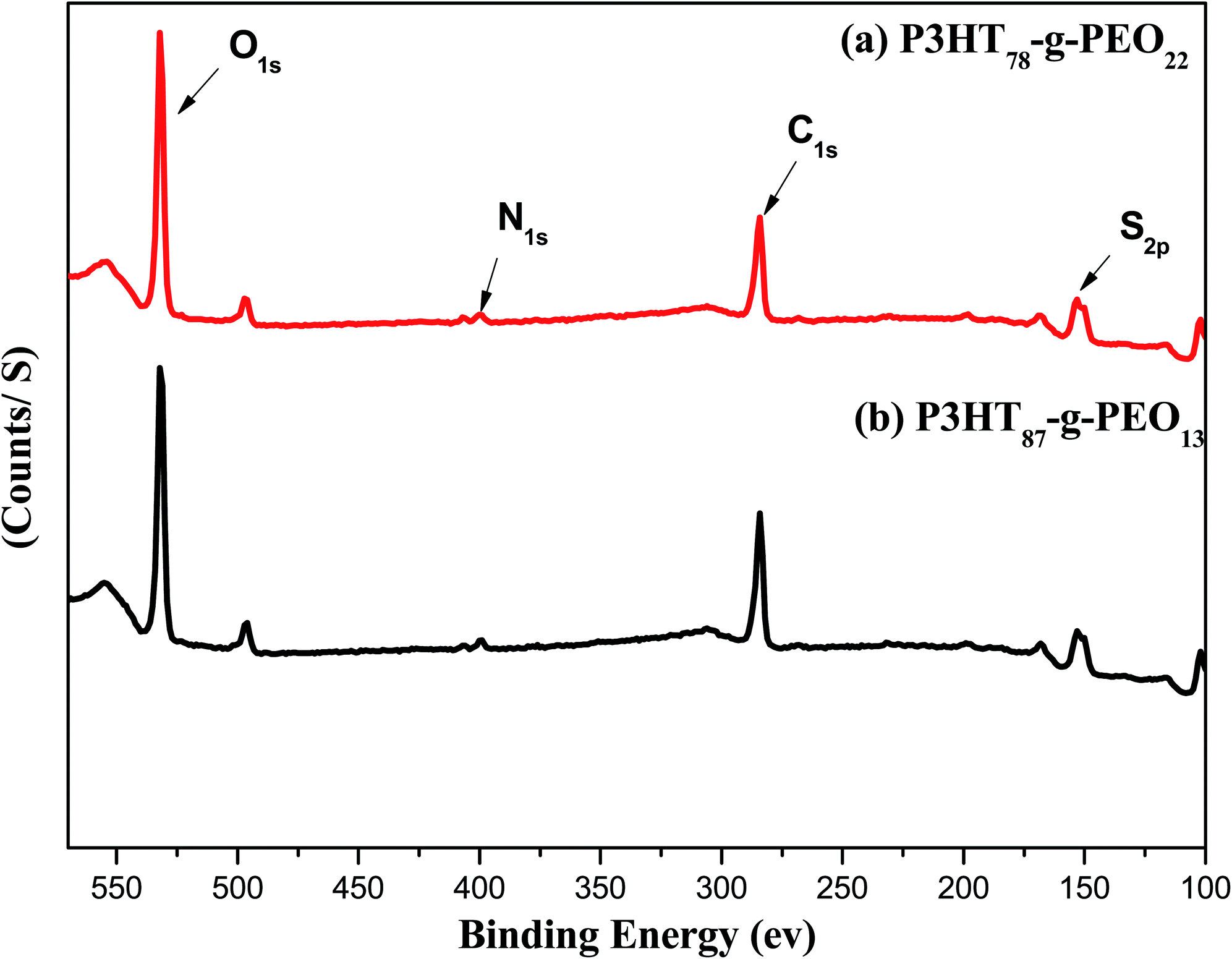
XPS survey spectra of the amphiphilic P3HT-g-PEO random copolymers (Fig. 3) revealed a signal related to the C1s orbital's at 284 eV, representing primarily the carbon atoms of the aromatic rings of the conjugated polymeric backbone;33 a signal for the N1s orbital's near 400 eV, assigned to the C–N bonds involving the nitrogen atoms of the triazole units;34 a signal for the S2p orbital's near 152 eV, representing the S atoms of the thiophene rings; and a signal for the O1s orbital's near 530 eV, representing the C–O–C units of the PEO blocks. These features are consistent with the successful attachment of PEO blocks to random P3HT copolymers.
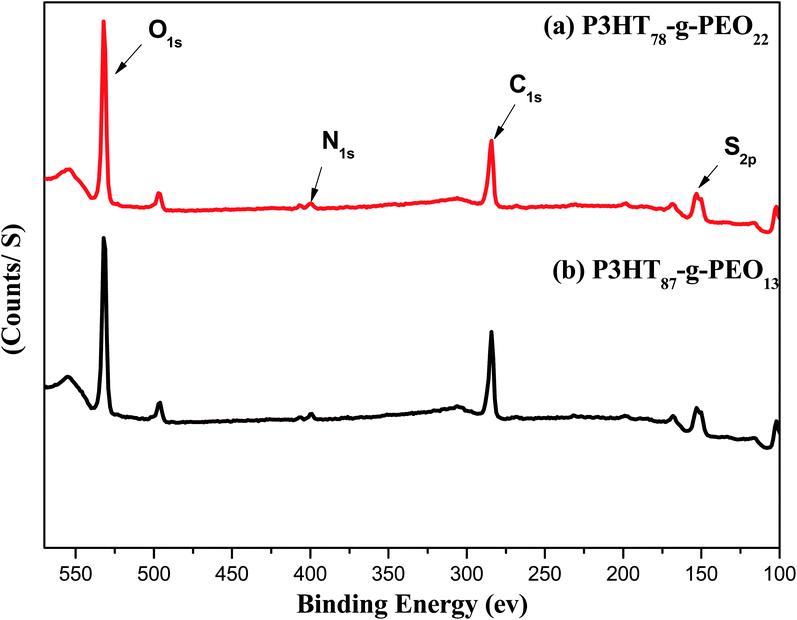 |
| | Fig. 3 XPS spectra of (a) P3HT78-g-PEO22 and (b) P3HT87-g-PEO13 random copolymers. | |
Thermal analyses of PT grafted PEO copolymers
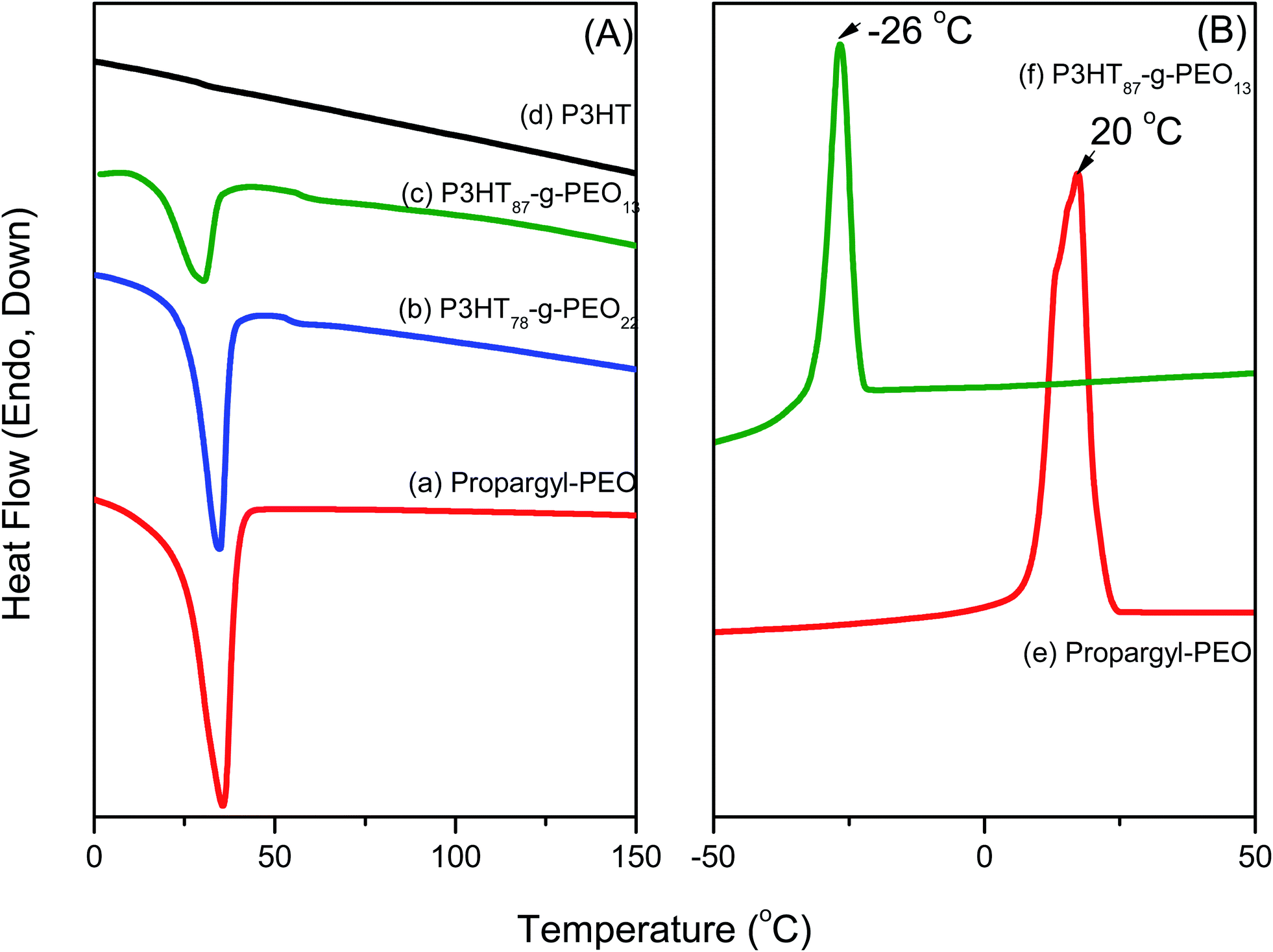
Fig. 4(a) presents DSC thermograms of random P3HT, PEO homopolymer, and two P3HT-g-PEO random copolymers, one featuring a large amount of PEO segments and the other low amount of segments, as determined using 1H NMR spectroscopy. During the second heating run of the random P3HT [Fig 4(a)], we observed neither a melting temperature (Tm) nor a glass transition temperature in the range 0–170 °C. PEO homopolymer exhibited a melting point at 35.6 °C, while the P3HT-g-PEO random copolymers featuring higher and lower ratios of PEO displayed melting temperatures of 34.5 and 30.6 °C, respectively. The observation of a single melting temperature, originating from the PEO segments, indicates that these PEO segments could form crystalline domains in the random copolymers as a result of phase separation. Fig. 4(b) presents DSC thermograms of PEO homopolymer and the P3HT-g-PEO random copolymer P3HT87-g-PEO13. During the cooling run, PEO exhibited a crystallization temperature near 20 °C. After we had used click chemistry to graft the PEO segments onto the random PT, the crystallization temperature shifted from +20 to −26 °C. Chen et al.35 reported that the degree of supercooling required to initiate crystallization in the lamellar microdomains (ΔT = 50 °C) is comparable to that associated with the PEO homopolymer, with exceedingly large undercooking required for crystallization in cylindrical micro domains 25 °C; (ΔT = 100 °C). This interestingly result also indicates that the microphase structure was formed as a result of a confinement effect in the P3HT-g-PEO random copolymer prepared in this study.36
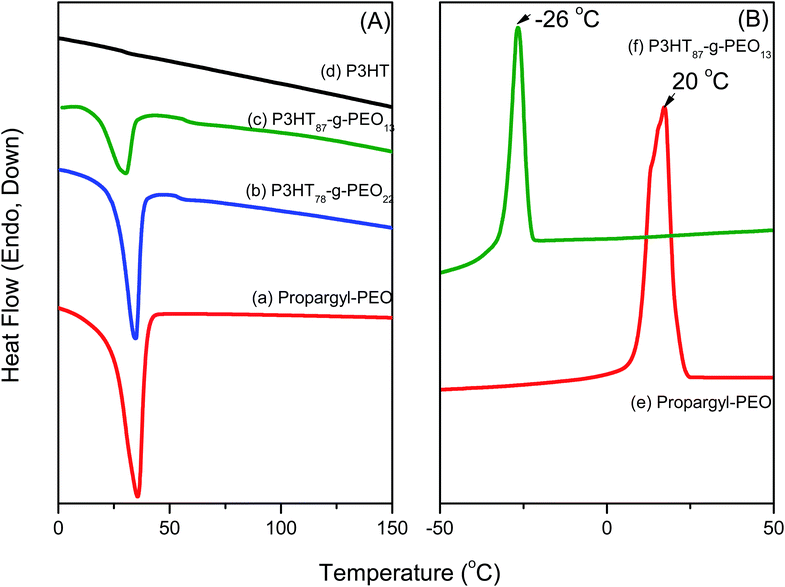 |
| | Fig. 4 (A) DSC traces (second heating run) of (a) PEO homopolymer, (b) P3HT78-g-PEO22 random copolymer, (c) P3HT87-g-PEO13 random copolymer, and (d) P3HT homopolymer. (B) DSC traces (first cooling run) of (e) PEO homopolymer and (f) P3HT87-g-PEO13 random copolymer. | |
UV-Vis and fluorescence spectra
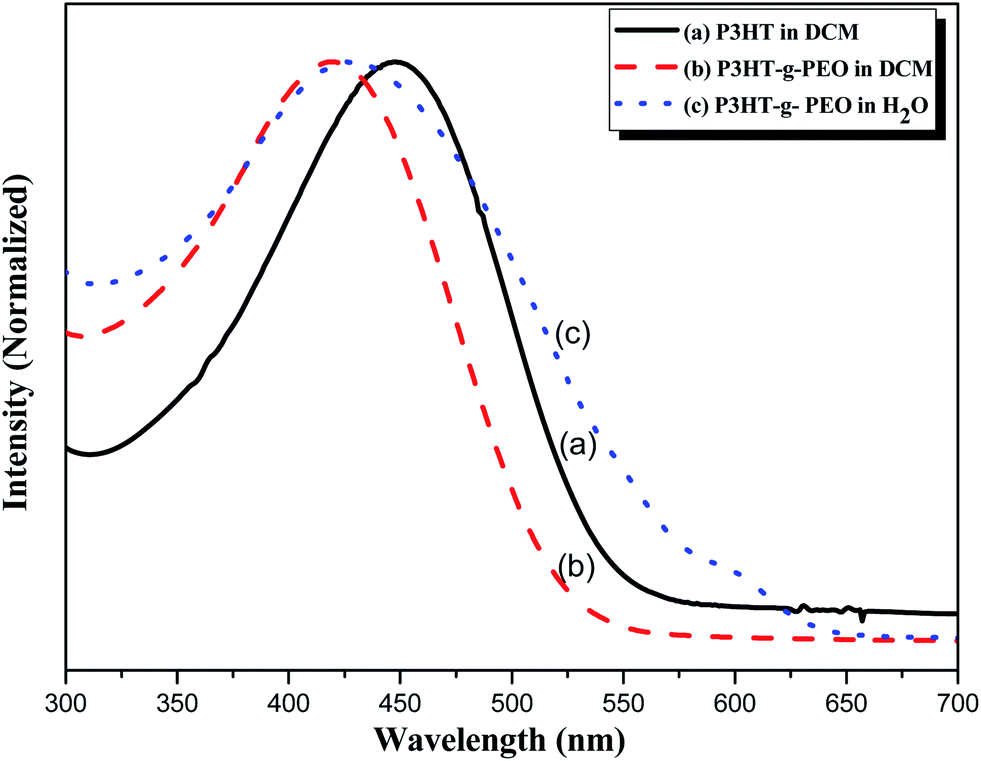
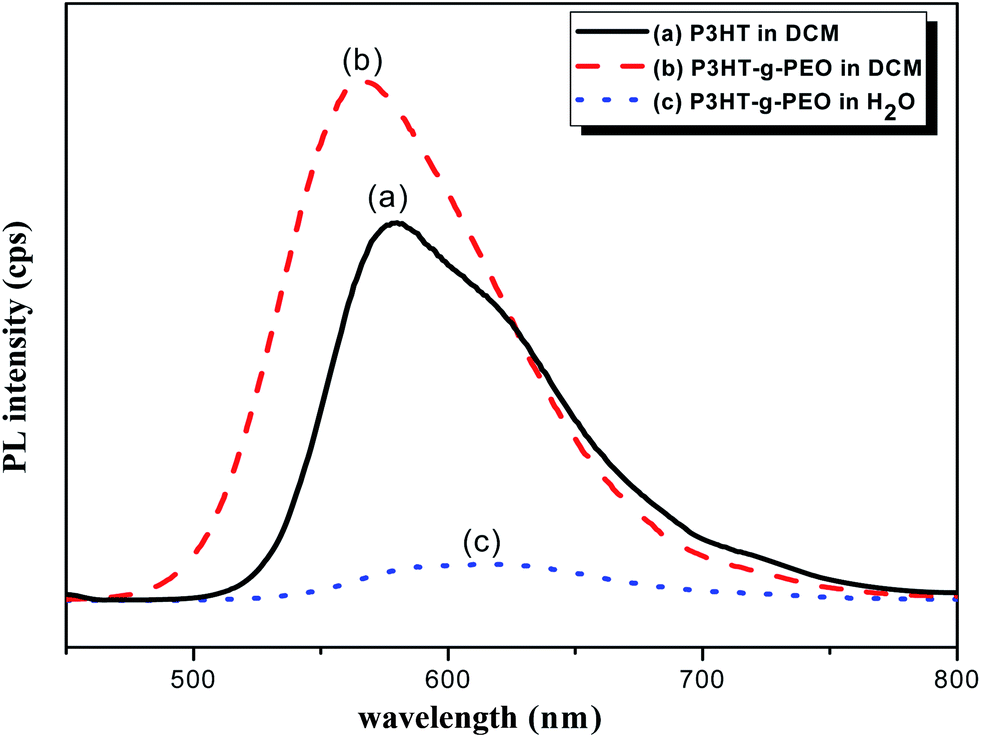
We used UV-Vis spectroscopy to examine the optical properties of the P3HT-g-PEO random copolymers. Because of their amphiphilic nature, we could disperse them in a wide range of solvents. In polar solvents, such as THF, DCM, and CHCl3, both the amphiphilic P3HT-g-PEO random copolymers were soluble and existed in the form of isolated chains. Fig. 5 displays the characteristic UV-Vis spectra of a P3HT homopolymer in a good solvent (DCM). The π–π* absorption peaks for P3HT and the P3HT-g-PEO random copolymer appear at 450 and 421 nm, respectively. These peaks are characteristic of regioregular P3HT in DCM,36 indicating that the attached PEO segments did not significantly affect the conformation of P3HT in this good solvent. When we dissolved the P3HT-g-PEO random copolymers in a selective solvent for the PEO segments, such as water and methanol, they organized into supramolecular assemblies, as evidenced by the red-shift in the signal for the π–π* absorption peaks in the UV-Vis spectra. Such a red-shifted absorption peak is characteristic of increased planarity of P3HT chains in a polymer assembly. Fig. 6 presents the fluorescence spectra of P3HT in DCM and the P3HT-g-PEO random copolymer in DCM and water. The fluorescence spectrum of P3HT in DCM featured a signal of high intensity at 576 nm, while that of the P3HT-g-PEO random copolymer exhibited a signal of high intensity at 565 nm in DCM, characteristic of its P3HT units. Thus, the fluorescence spectra confirmed the results from the UV-Vis spectra: that the attachment of PEO segments did not affect the conformation of P3HT in the good solvent. The fluorescence intensity of P3HT-g-PEO random copolymer was quenched in water, a good solvent for the PEO blocks; this finding is also indicative of tightly packed P3HT chains and strong interchain coupling in its polymer assemblies.37–39
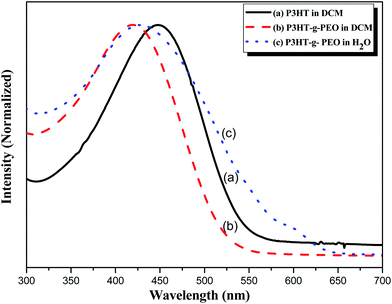 |
| | Fig. 5 UV-Vis spectra of (a) P3HT homopolymer in DCM and (b and c) P3HT78-g-PEO22 random copolymer in (b) DCM and (c) H2O. | |
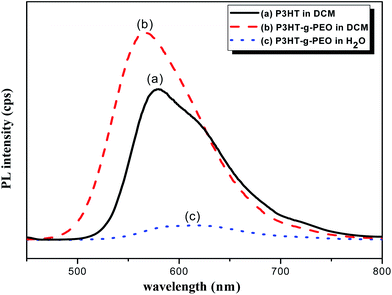 |
| | Fig. 6 PL spectra of (a) P3HT homopolymer in DCM and (b and c) P3HT78-g-PEO22 random copolymer in (b) DCM and (c) H2O. | |
Self-assembled structures formed from P3HT-g-PEO random copolymers in solution
Our amphiphilic P3HT-g-PEO random copolymers could self-assemble into micelles in aqueous solution; we employed the fluorescence probe method to analyze their critical micellar concentrations (CMCs), using pyrene a probe of the photo physical properties. To a series of ampoules, we added 50 μL of a solution of pyrene in acetone and then evaporated the acetone under reduced pressure. We then added aqueous solutions (5 mL) containing different concentrations (from 333 × 10−3 to 12.52 × 10−3 mg L−1) of the amphiphilic random copolymers to the ampoules containing the residue. The concentration of pyrene was fixed at 6 × 10−6 M to ensure its solubilization equilibrium. We sonicated the aqueous solutions of the amphiphilic P3HT-g-PEO random copolymers for 10 min and then stirred them for 24 h at room temperature. Upon excitation at 450 nm, Fig. 7(A) and (B) display the emission spectra recorded in the range from 450 to 800 nm. The fluorescence intensity underwent an abrupt increase upon increasing the concentration of the P3HT-g-PEO random copolymers [Fig. 7(C)], indicating the formation of micelles and the transfer of pyrene into the hydrophobic cores of these micelles. The CMCs of the two amphiphilic P3HT78-g-PEO22 and P3HT87-g-PEO13 tested in this study were 2.23 × 10−3 and 1.74 × 10−3 g mL−1, respectively. 1H NMR spectroscopy provided further evidence for the micellization of our amphiphilic P3HT-g-PEO random copolymers in aqueous solution. The 1H NMR spectrum of the micelles in D2O [Fig. 7(D)] exhibited two apparent signals at 4.63 and 3.48 ppm, which we assign to the hydrogen atoms of the CH2O groups in the PEO units and the solvent peak (HOD), respectively. The signals of the protons in the P3HT segment disappeared completely in D2O, unlike the situation for the copolymers in CDCl3, suggesting that the amphiphilic P3HT-g-PEO random copolymers formed core/shell micellar structures featuring an isolated hydrophobic inner core and a hydrophilic outer shell.40 Because P3HT blocks are hydrophobic and water-insoluble, but PEO blocks are hydrophilic and water soluble we expected these amphiphilic random copolymers to undergo self-assembly into nanosized micelles in selective solvent systems. As a general procedure, we first dissolved the amphiphilic random copolymers under study in a good solvent for both blocks (typical concentration: 1 mg mL−1) and then added a second, non-selective solvent a poor solvent for one of the blocks to the polymer solution very slowly.
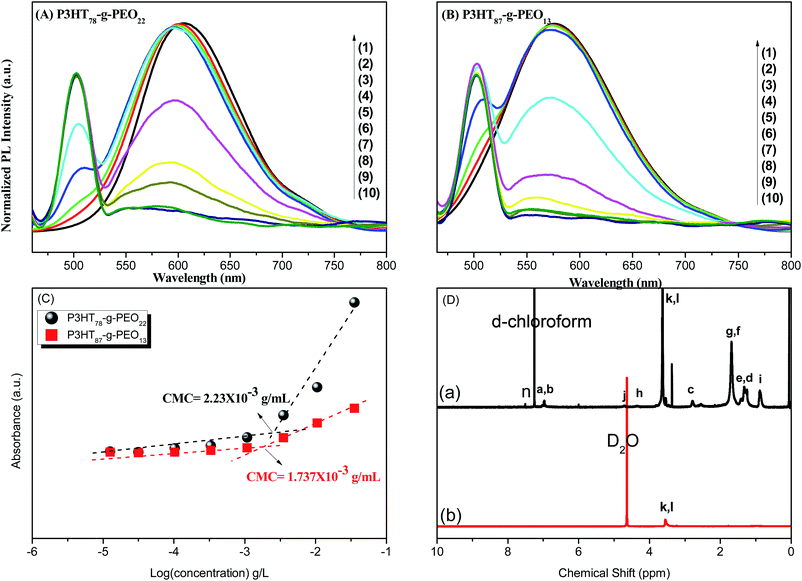 |
| | Fig. 7 (A and B) Emission spectra of pyrene at concentrations of (1) 333, (2) 111, (3) 34.2, (4) 10.5, (5) 3.51, (6) 1.07, (7) 0.33, (8) 0.102, (9) 0.03, and (10) 12.52 × 10−3 mg L−1 for (A) P3HT78-g-PEO22 and (B) P3HT87-g-PEO13 random copolymers. (C) Intensity ratio I3/I1 in the emission spectra plotted with respect to the logarithm of the polymer concentration. (D) 1H NMR spectra of P3HT78-g-PEO22 random copolymer in (a) CDCl3 and (b) D2O. | |
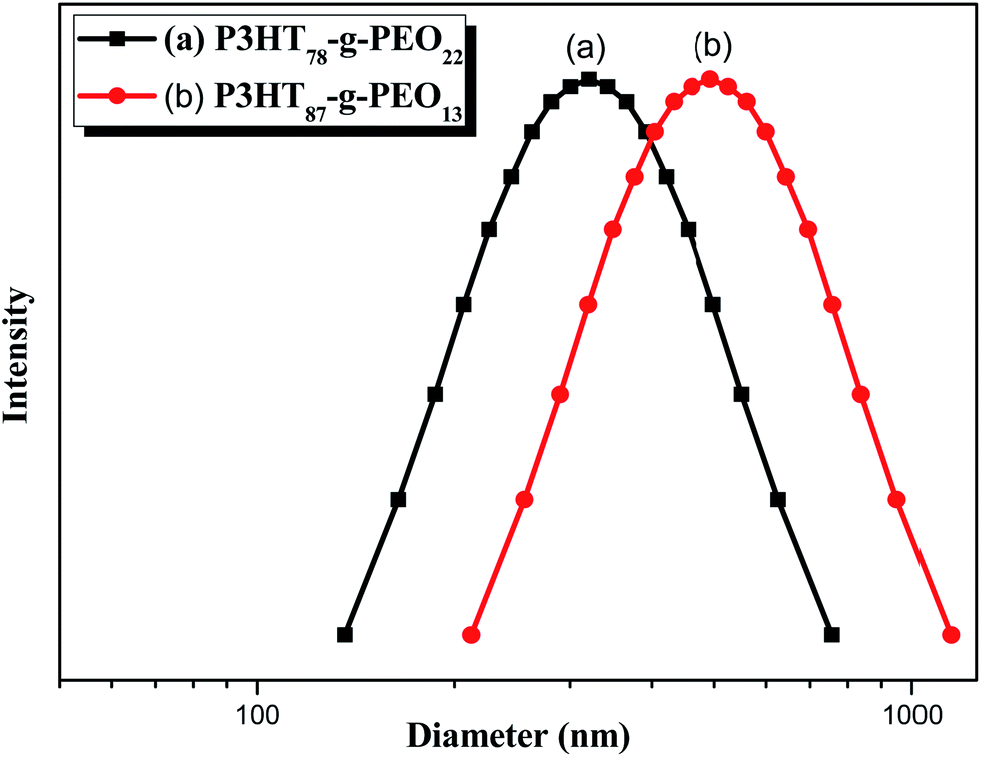
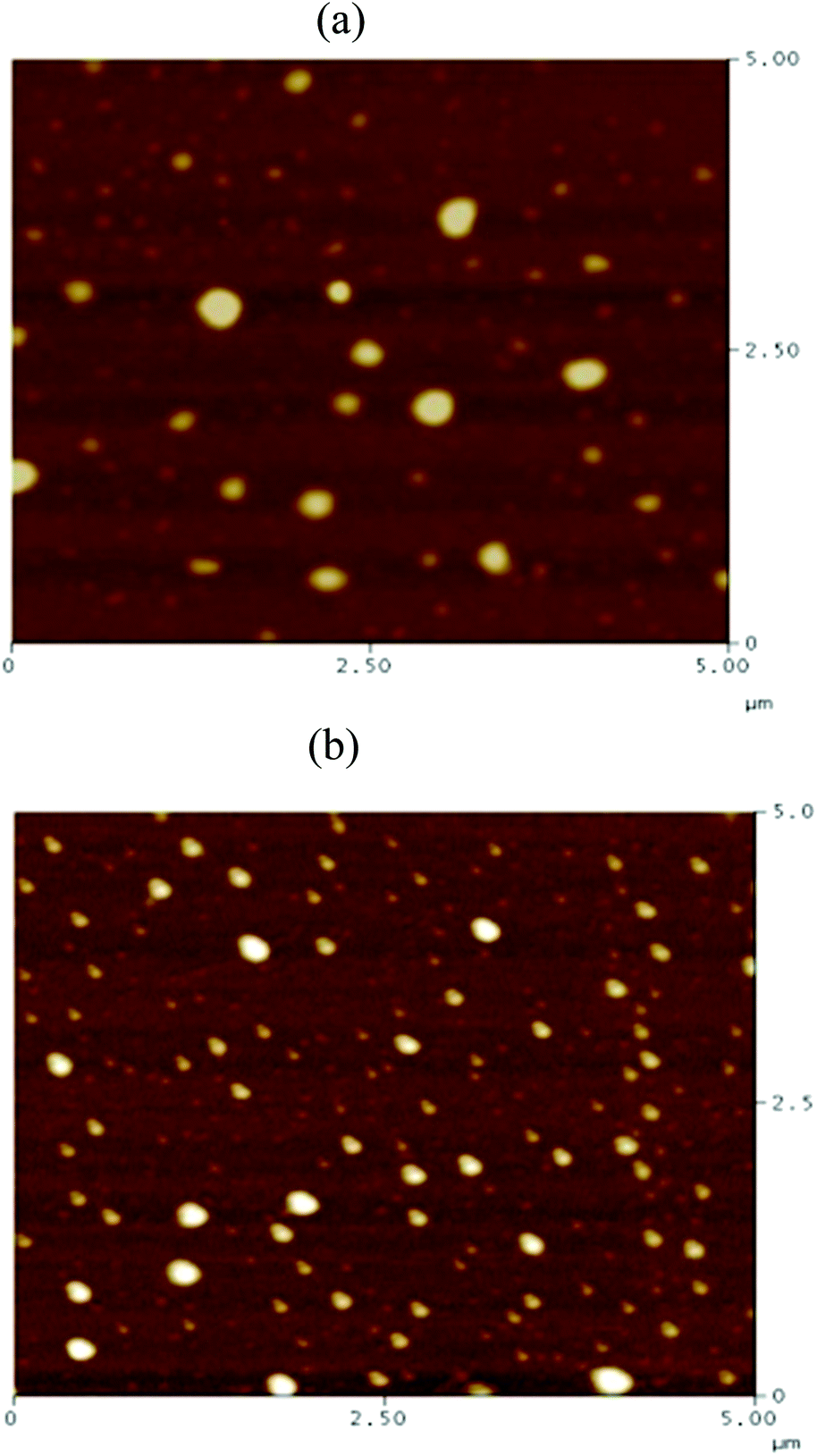
Recently, Cheng et al. systematically studied the micellization and morphological transitions of a PS-b-PEO block copolymer in DMF–water and DMF–MeCN systems.41 They found that the micellar morphologies were strongly dependent on the water and MeCN contents and on the polymer concentration. Here, we chose a different selective solvent (water) to study the effects of solvent on the self-assembly of the amphiphilic random copolymers. First, we dissolved the P3HT-g-PEO random copolymer in a common solvent (DMF) and then added the selective solvent (H2O) slowly to induce phase separation and aggregation of the P3HT blocks, which condensed gradually with the PEO blocks constituting the outer shells of the particles. Fig. 8 presents a set of TEM images of the micelles formed from two P3HT-g-PEO random copolymers at an initial concentration of 0.5 mg mL−1 in the DMF–H2O system. Spherical micelles were formed; these spheres were all nearly identical in size (ca. 60–79 nm). At the highest concentration of H2O (9 wt%), these spherical nanoparticles were closely packed and had very uniform size. We also expected the mobility of the P3TT blocks to be severely restricted, because H2O is a poor solvent for P3HT; thus, we predicted that the morphology might not change at a higher concentration of water We obtained DLS data to study the hydrodynamic diameters (Dh) of the micelles (Fig. 9). We observed only a single peak associated with the polymer aggregates as signals near 320.7 and 492.4 nm for our two amphiphilic random polythiophenes P3HT78-g-PEO22 and P3HT87-g-PEO13, respectively providing direct evidence for micelle formation. The diameters of these micelles, as measured from TEM images, were within the range 60–79 nm; these values are smaller than those measured using DLS, presumably because the preparation of the samples for TEM observation involved evaporation of the micelle particles, making chain collapse and micelle shrinkage unavoidable.42 Fig. 10 presents AFM images of the two amphiphilic random P3HT-g-PEO systems in water—a selective solvent for the PEO segments. Our amphiphilic polythiophenes (P3HT78-g-PEO22 and P3HT87-g-PEO13) also formed spherical structures in these aqueous systems, with diameters of 117.24 and 242.5 nm, respectively.
 |
| | Fig. 8 TEM images of (a) P3HT78-g-PEO22 and (b) P3HT87-g-PEO13 in DMF–H2O containing 5 wt% H2O and of (c) P3HT87-g-PEO13 in DMF–H2O containing 9 wt% H2O. | |
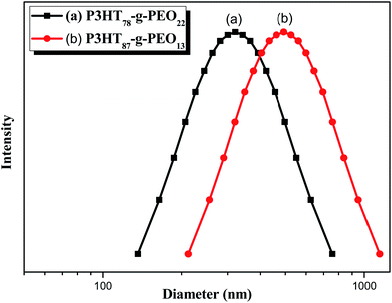 |
| | Fig. 9 DLS data for (a) P3HT78-g-PEO22 and (b) P3HT87-g-PEO13 random copolymers in DMF–H2O containing 9 wt% H2O. | |
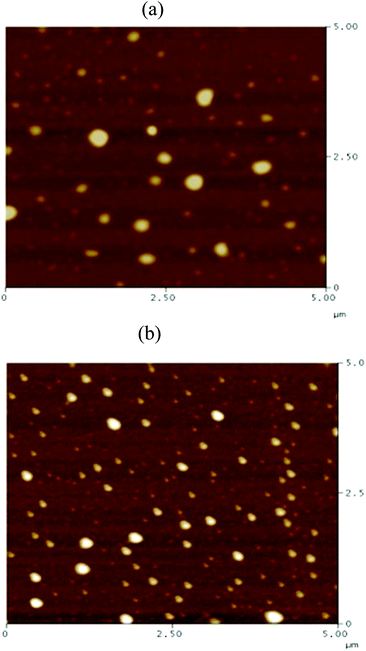 |
| | Fig. 10 AFM images of (a) P3HT78-g-PEO22 and (b) P3HT87-g-PEO13 random copolymers deposited on a silica substrate from aqueous solutions at 1 mg mL−1. | |
Conclusions
We have successfully prepared P3HT-g-PEO random copolymers, possessing hydrophobic PT backbones and hydrophilic PEO side chains, through oxidative polymerization and click chemistry. Optical data from UV-Vis and photoluminescence spectroscopy supported the expected structures of these random P3HT-g-PEO copolymers. Interestingly, these copolymers could form micelles in aqueous solutions, with CMCs as low as 1.7–2.3 × 10−3 mg L−1 and spherical particle diameters of approximately 60–75 nm (based on TEM imaging). We suspect that such P3HT polymers presenting PEO units on their side chains will be promising materials for a variety of bioengineering and biomedical applications.
Acknowledgements
This study was supported financially by the National Science Council, Taiwan, Republic of China, under contracts NSC 100-2221-E-110-029-MY3 and NSC 100-2628-E-110-001. Authors thank Mr Hsien-Tsan Lin of Regional Instruments Center at National Sun Yat-Sen University for his help in TEM experiments.
References
- A. Schenning and E. W. Meijer, Chem. Commun., 2005, 26, 3245 RSC.
- M. He, W. Han, J. Ge, Y. Yang, F. Qiu and Z. Lin, Energy Environ. Sci., 2011, 4, 2894 CAS.
- M. R. Raj and S. Anandan, RSC Adv., 2013, 3, 14595 RSC.
- C. L. Liu, C. H. Lin, C. C. Kuo, S. T. Lin and W. C. Chen, Prog. Polym. Sci., 2011, 36, 603 CrossRef CAS PubMed.
- Y. F. Tao, B. McCulloch, S. Kim and R. A. Segmalman, Soft Matter, 2009, 5, 4219 RSC.
- S. K. Patra, E. Ahmed, G. R. Whittell, D. J. Lunn, E. L. Dunphy, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8842 CrossRef CAS PubMed.
- J. Peng, Y. C. Han, W. Knoll and D. H. Kim, Macromol. Rapid Commun., 2007, 28, 1422 CrossRef CAS.
- C. R. Craley, R. Zhang, T. Kowalewski, R. D. McCullough and M. C. Stafen, Macromol. Rapid Commun., 2009, 30, 11 CrossRef CAS PubMed.
- H. S. O. Chan and S. C. Ng, Prog. Polym. Sci., 1998, 23, 1167 CrossRef CAS.
- G. Zotti, R. A. Marin and M. C. Gallazzi, Chem. Mater., 1997, 9, 2945 CrossRef CAS.
- S. H. Hosseini and A. Entezami, Polym. Adv. Technol., 2001, 12, 524 CrossRef CAS.
- I. Osaka and R. D. McCullough, Acc. Chem. Rev., 2008, 41, 1202 CrossRef CAS PubMed.
- A. R. Aiyar, J. I. Hong, R. Nambiar, D. M. Collaard and E. R. Manis, Adv. Funct. Mater., 2011, 21, 2652 CrossRef CAS.
- T. Ghoos, Q. Malinkiewicz, B. Conings, L. Lutsen, D. J. Vanderzande, H. J. Bolink and W. Maes, RSC Adv., 2013, 3, 25197 RSC.
- M. He, W. Han, J. Ge, W. J. Yu, Y. L. Yang, F. Qiu and Z. Q. Lin, Nanoscale, 2011, 3, 3159 RSC.
- T. Shiraki, A. Dawn, Y. Tsuchiya and S. Shinkai, J. Am. Chem. Soc., 2010, 132, 13928 CrossRef CAS PubMed.
- C. Dai, W. C. Yen, Y. H. Lee, C. C. Ho and W. F. Su, J. Am. Chem. Soc., 2007, 129, 11036 CrossRef CAS PubMed.
- Y. J. Lee, S. H. Kim, H. Yang, M. Jang, S. S. Hwang, H. S. Lee and K. Y. Baek, J. Phys. Chem. C, 2011, 115, 4228 CAS.
- H. C. Moon, A. Anthonusamy, J. K. Kim and A. Hirao, Macromolecules, 2011, 44, 1894 CrossRef CAS.
- B. W. Boudouris, C. D. Frisbie and M. A. Hillmyer, Macromolecules, 2008, 41, 67 CrossRef CAS.
- V. Ho, B. W. Boudouris, B. L. McCulloch, C. G. Shuttle, M. Bukhardt, M. L. Chabinyc and R. A. Segalman, J. Am. Chem. Soc., 2011, 133, 9270 CrossRef CAS PubMed.
- S. J. Park, S. G. Kang, M. Fryd and J. G. Saven, J. Am. Chem. Soc., 2010, 132, 9931 CrossRef CAS PubMed.
- Z. J. Gu, T. Kanto, K. Tsuchiya, T. Shimomura and K. Ogino, J. Polym. Sci., Part A-1: Polym. Chem., 2011, 49, 2645 CrossRef CAS.
- E. Kaul, V. Senkovskyy, R. Tkachov, V. Bocharova, H. Komber, M. Stamm and A. Kiriy, Macromolecules, 2010, 49, 77 CrossRef.
- M. Urien, H. Erothu, E. Cloutet, R. C. Hiorns, L. Vignau and H. Cramail, Macromolecules, 2008, 41, 7033–7040 CrossRef CAS.
- Z. Q. Wu, R. J. One, Z. Chen, Z. C. Li and C. W. Bielawski, Polym. Chem., 2011, 2, 300 RSC.
- X. C. Pang, L. Zhao, C. W. Feng and Z. Q. Lin, Macromolecules, 2011, 44, 7176 CrossRef CAS.
- Z. C. Li, R. J. Ono, Z. Q. Wu and C. W. Bielawski, Chem. Commun., 2011, 47, 197 RSC.
- A. C. Kamps, M. Fryd and S. J. Park, ACS Nano, 2012, 6, 2844 CrossRef CAS PubMed.
- S. N. Patel, A. E. Javier, G. M. Stone, S. A. Mullin and N. P. Balsara, ACS Nano, 2012, 6, 1589 CrossRef CAS PubMed.
- Z. Lei, L. P. Richard, L. Z. Karen, K. S. Kristoferr and R. D. McCullough, Macromolecules, 2003, 36, 61 CrossRef.
- L. H. Tu, Y. F. Tsai and Y. C. Mou, J. Mater. Chem., 2008, 18, 1771 RSC.
- I. F. Venditti, C. Palazzesi, P. Prosposto, M. Casalboni, C. Cametti, C. Battocchio, G. Polzonetti and M. V. Russo, J. Colloid Interface Sci., 2010, 348, 424 CrossRef CAS PubMed.
- A. S. Sarac, S. A. M. Tofail, M. Serantoni, J. Henery, V. J. Cunnane and J. B. McMonagle, Appl. Surf. Sci., 2004, 222, 148 CrossRef CAS PubMed.
- H. L. Chen, S. C. Hsiao, T. L. Lin, K. Yanauchi, H. Hasegawa and T. Hashimoto, Macromolecules, 2001, 34, 671–674 CrossRef CAS.
- J. G. Li, Y. D. Lin and S. W. Kuo, Macromolecules, 2011, 44, 9295–9309 CrossRef CAS.
- G. L. Tu, H. B. Li, M. Forester, R. Heiderhoff, L. J. Balk, R. Sigel and U. Scherf, Small, 2007, 3, 1001 CrossRef CAS PubMed.
- S. Samitsu, T. Shimomura and K. Ito, Macromolecules, 2008, 41, 8000 CrossRef CAS.
- A. C. Kamps, M. Helen, M. Cativo, M. Fryd and S. J. Park, Macromolecules, 2014, 47, 161 CrossRef CAS.
- S. W. Kuo, H. F. Lee, C. F. Huang, C. J. Huang and F. C. Chang, J. Polym. Sci., Part A-1: Polym. Chem., 2008, 46, 3108 CrossRef CAS.
- P. Bhargava, J. X. Zheng, P. Li, R. P. Quirk, F. W. Harri and S. Z. D. Cheng, Macromolecules, 2006, 39, 4880 CrossRef CAS.
- P. C. Li, Y. C. Lin, M. Chen and S. W. Kuo, Soft Matter, 2013, 9, 11257 RSC.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.