Enhanced electrochemical response for mercury ion detection based on poly(3-hexylthiophene) hybridized with multi-walled carbon nanotubes
Shaojun Yangab,
Dongli Mengb,
Jinhua Sunb,
Wenpeng Houb,
Yangbin Dingb,
Shidong Jiangb,
Yan Huang*a,
Yong Huangb and
Jianxin Geng*b
aKey Laboratory of Oil & Gas Fine Chemicals, Ministry of Education & Xinjiang Uyghur Autonomous Region, Xinjiang University, Urumqi 830046, China. E-mail: xindhuangyan@yahoo.cn
bTechnical Institute of Physics and Chemistry, Chinese Academy of Sciences, 29 Zhongguancun East Road, Haidian District, Beijing 100190, China. E-mail: jianxingeng@mail.ipc.ac.cn; Fax: +86-10-6255 4670; Tel: +86-10-8254 3416
First published on 25th April 2014
Abstract
In this study, multi-walled carbon nanotubes (MWNTs) were covalently modified by grafting poly(3-hexylthiophene) (P3HT) on to their surfaces. The modified MWNTs (MWNT-g-P3HT) showed enhanced dispersibility in common solvents, such as THF. Due to the intimate interaction between the P3HT and MWNTs, the MWNT-g-P3HT showed improved miscibility with P3HT, and the composite of the MWNT-g-P3HT and P3HT (MWNT-g-P3HT@P3HT) was likely to form continuous films. Notably, the MWNT-g-P3HT@P3HT composite films demonstrated an enhanced electrochemical response for the quantitative detection of Hg2+ ions, due to the synergistic effect of the electrocatalytic properties from the MWNTs and P3HT.
Introduction
As a type of typical conjugated polymer, polythiophene and its derivatives have attracted enormous attention due to their wide application in areas such as electronics,1 optoelectronics,2,3 and detection techniques.4 In recent years, great progress has been made in the chemical detection technique utilising polythiophene derivatives, which has been extensively applied for the detection of small ions,5–7 hazardous organic molecules,8–10 oxidizing vapours,11 and biomolecules, including DNA, amino acids, proteins, and microbial particles.12–23 Basically, chemical detection using polythiophenes is based on the transformation of chemical interactions between the conjugated polymers and the analytes, into easily measured optical or electrical signals. Water-soluble polythiophene derivatives are an important class of conjugated polymers for fluorescence detection. The fluorescence properties of the polythiophene derivatives are sensitive to the conformation and the aggregation of the polymers, which commonly reflect the electrostatic or chelation interactions between the analytes and the designed functional groups in the side chains.6,22,23 On the other hand, electrochemical sensors which utilise polythiophenes are commonly designed by linking a probe and a label onto the main chains of the polythiophenes, or physically absorbing the probe and the label on the surface of the polythiophene films.14–17 The interaction between the label and the analyte can cause changes in the electrochemical signal of the probe. This technique has been widely used for DNA detection, for example, by using single-strand DNA as the label, and ferrocene as the probe.15,16Hybridized electrodes of polythiophenes also possess unique features derived from the advantages of both the polythiophene and the nanofillers. Carbon nanotubes (CNTs) have been demonstrated to be a type of important filler, and impart their features, such as excellent electrocatalytic activity, enhanced sensitivity, and electrical conductivity, to the resultant composites.24,25 In addition, CNT/polythiophene electrodes also show a synergistic effect of the electrocatalytic properties from both the CNTs and the conjugated polymers, leading to an enhanced electrochemical response of the hybridized electrodes compared to that of the non-hybridized counterpart.26 However, agglomeration of CNTs is likely to take place in CNT–conjugated polymer composite films, even those with a low loading of CNTs, leading to unexpected interfaces between the two components. In this case, the features of the CNTs may not be fully exhibited. Therefore, research work on the surface modification of CNTs is still required to enhance the miscibility between the CNTs and corresponding conjugated polymers, and thereafter to improve the application performance of the CNT–conjugated polymer composites.
In this research, we covalently grafted P3HT on to the surface of multi-walled carbon nanotubes (MWNTs), by taking advantage of the amidation reaction between the carboxylic acid groups on the surface of the MWNTs, and the amine groups of an amino-terminated P3HT. The modified MWNTs were designated as MWNT-g-P3HT. Due to the intimate interactions between the MWNTs and P3HT, MWNT-g-P3HT showed enhanced dispersibility when mixed with P3HT. The resultant composite was named MWNT-g-P3HT@P3HT. Significantly, the MWNT-g-P3HT@P3HT composite films exhibited an enhanced electrochemical response for the detection of Hg2+ ions.
Experimental section
Materials
MWNTs of ca. 8 nm diameter were purchased from Chengdu Organic Chemicals Co. Ltd. Before they were used, the MWNTs were purified by dry oxidization at 300 °C for 60 min under a flow of dry air (0.1 SLM), and washing in 12 N HCl solution. Hg(NO3)2·H2O (extra purity grade) was purchased from Sinopharm Chemical Co. Ltd.Synthesis of MWNT-g-P3HT
As shown in Fig. 1a, an amino-terminated P3HT (P3HT-NH2) was polymerized using the Grignard metathesis reaction,27 which is a universal method for the synthesis of polythiophenes with a narrow molecular weight distribution and high regularity. Amine groups were introduced into the polymer chains by terminating the polymerization using a proper Grignard reagent.28 The number average molecular weight of the P3HT-NH2 was determined to be ca. 3200 by gel permeation chromatography using a polystyrene standard. | ||
| Fig. 1 Routes for the synthesis of (a) P3HT-NH2 and (b) MWNT-g-P3HT. | ||
MWNT-g-P3HT was synthesized through the condensation of the carboxylic acid groups on the surface of the MWNTs and the amine groups of the P3HT-NH2 (Fig. 1b). In a typical reaction, purified MWNTs (21 mg) were dispersed in DMF, with the aid of sonication, for 3 h. To the suspension, SOCl2 (21 mL) was added, and this was refluxed at 70 °C for 36 h. The product was obtained by centrifuge, and purified by 4 cycles of washing with anhydrous THF and centrifugation. To carry out the amidation reaction, the MWNT suspension in anhydrous THF (30 mL) was added to a P3HT-NH2 (200 mg) solution in anhydrous THF (50 mL), followed by the slow addition of triethylamine (3 mL) at 0 °C. The reaction was allowed to take place at 50 °C for 48 h. Finally, MWNT-g-P3HT was obtained after cycles of washing with THF and centrifugation until the supernatant became colourless.
Electrochemical detection of Hg2+ ions
Electrochemical detection of Hg2+ ions was performed using the MWNT-g-P3HT@P3HT composite films. To prepare the electrochemically active electrodes, MWNT-g-P3HT@P3HT composite suspensions with different ratios of MWNT-g-P3HT to P3HT (0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1, 1.5:1, 2:1, and 2.5:1) were prepared using the MWNT-g-P3HT and pure P3HT, where the concentration of pure P3HT was ca. 0.5 mg mL−1 in the resultant suspensions. Finally, MWNT-g-P3HT@P3HT films were prepared on the tip of glassy carbon (GC) electrodes by casting 8 μL of the composite suspensions, followed by natural drying.
:1, 1:1, 1.5:1, 2:1, and 2.5:1) were prepared using the MWNT-g-P3HT and pure P3HT, where the concentration of pure P3HT was ca. 0.5 mg mL−1 in the resultant suspensions. Finally, MWNT-g-P3HT@P3HT films were prepared on the tip of glassy carbon (GC) electrodes by casting 8 μL of the composite suspensions, followed by natural drying.
Electrochemical detection of Hg2+ ions was performed by cyclic voltammetry (CV) using a three-electrode configuration, where an Ag/AgCl electrode was used as the reference electrode, a Pt electrode was used as the counter electrode, the MWNT-g-P3HT@P3HT composite films coated on a GC electrode was used as the working electrode, and a KNO3 solution (0.2 M) was used as the supporting electrolyte. Hg(NO3)2·H2O was used as the Hg2+ ion source.
Characterization
FT-IR spectra were recorded on an Excalibur 3100 spectrometer with a resolution of 0.2 cm−1 using KBr pellets. Raman spectra were collected on a Renishaw inVia-Reflex confocal Raman microscope with an excitation wavelength of 532 nm. Thermogravimetric analysis (TGA) was performed with a Q50 TGA at a scanning rate of 5 °C min−1, under the protection of N2. Transmission electron microscopy (TEM) observations were performed on a JEOL JEM-2100 transmission electron microscope operated at 200 kV. TEM samples were prepared by dropping the suspension of purified MWNTs in DMF, or the suspension of the MWNT-g-P3HT in THF, on 400 mesh Cu grids with supporting carbon film. TEM samples of P3HT film and MWNT-g-P3HT@P3HT composite film were prepared on the amorphous carbon films coated on mica slides, by spin coating (800 rpm) the solution or suspension (ca. 1 mg mL−1), followed by the transfer of the films onto Cu grids.29 CV curves were collected on a Zennium 40088 electrochemical workstation using a three-electrode configuration.Results and discussion
To enhance the miscibility of CNTs and polymers, surface modification of the CNTs is commonly used.30,31 In this process, the attributes of the two components can also be combined in the resultant composites. Noncovalent and covalent strategies are the two major approaches for surface modification of CNTs. The noncovalent approach can effectively preserve the conjugated structures on the surface of the CNTs,30 whereas the covalent approach leads to a more intimate and stabilized connectivity between the CNTs and the polymers, resulting in strong electronic interactions.31 Fig. 1b shows the route for the covalent grafting of P3HT on to the surface of MWNTs, which takes advantage of the amidation reaction between the carboxylic acid groups on the surface of the oxidized MWNTs, and the amine groups of the P3HT-NH2.The MWNT-g-P3HT and the intermediate products in the grafting process were characterized using FT-IR spectroscopy (Fig. 2). Compared to the FT-IR spectrum of the purified MWNTs, the oxidized MWNTs yield a spectrum which contains enhanced peaks at 1713 and 1381 cm−1, corresponding to the vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O–H in the carboxylic acid groups, respectively. Upon the amidation reaction, strong vibration peaks were found at 1654, 1628, and 1584 cm−1, corresponding to the amide in the spectrum of the MWNT-g-P3HT. This finding indicates the transformation of the carboxylic acid groups into amides. In addition, we can also find clear vibration peaks from the C–H bond at ca. 2900 cm−1, which is due to the C–H bonds in the hexyl groups of the grafted P3HT.
O and C–O–H in the carboxylic acid groups, respectively. Upon the amidation reaction, strong vibration peaks were found at 1654, 1628, and 1584 cm−1, corresponding to the amide in the spectrum of the MWNT-g-P3HT. This finding indicates the transformation of the carboxylic acid groups into amides. In addition, we can also find clear vibration peaks from the C–H bond at ca. 2900 cm−1, which is due to the C–H bonds in the hexyl groups of the grafted P3HT.
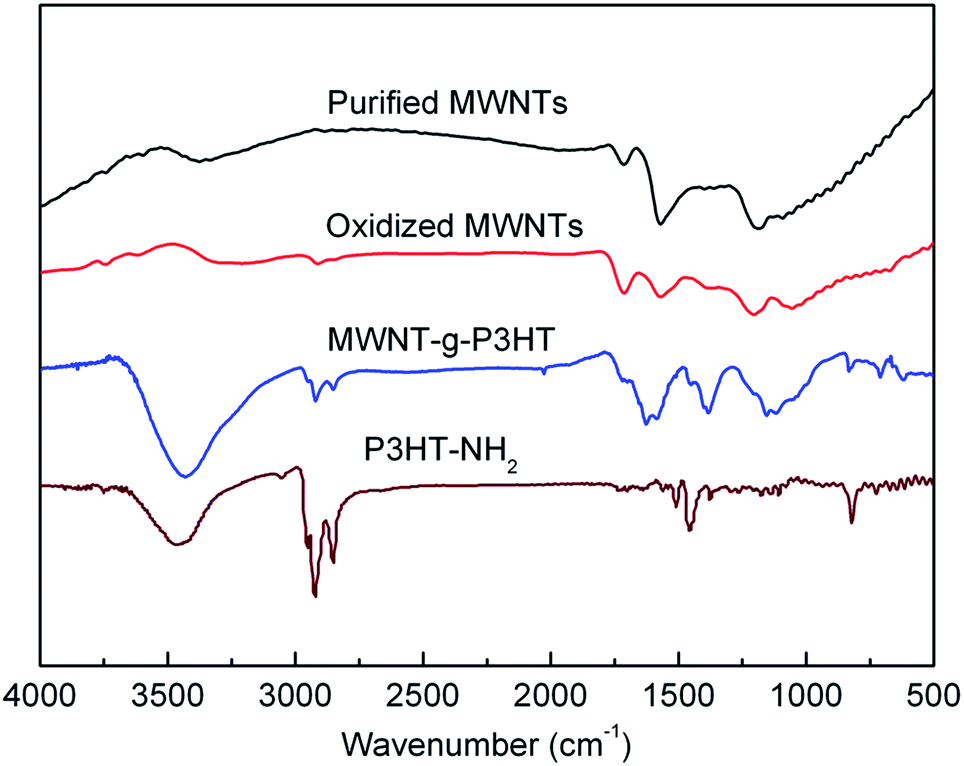 | ||
| Fig. 2 FT-IR spectra of the purified MWNTs, oxidized MWNTs, P3HT-NH2, and the MWNT-g-P3HT. | ||
The thermal stability and composition of the MWNT-g-P3HT were investigated using TGA (Fig. 3a). The purified MWNTs are thermally stable up to 900 °C, as no obvious mass loss was detected below this temperature. The oxidized MWNTs show a mass loss before 400 °C, which might be due to thermal degradation of the functional groups and small carbonaceous fragments formed during the oxidation. P3HT shows a marked mass loss between 400 and 500 °C. As expected, MWNT-g-P3HT shows features of both the oxidized MWNTs and the P3HT: a gradual mass loss before 400 °C, a small rapid mass loss between 400 and 500 °C, and a relatively stable period up to 800 °C. Thus, the content of P3HT in the MWNT-g-P3HT can be estimated to be 10.5%.
 | ||
| Fig. 3 (a) TGA curves for the purified MWNTs, oxidized MWNTs, P3HT-NH2, and the MWNT-g-P3HT; (b) Raman spectra of the purified MWNTs, oxidized MWNTs, P3HT-NH2, and the MWNT-g-P3HT. | ||
To further characterize the interface interactions between the MWNTs and P3HT in the MWNT-g-P3HT, Raman spectroscopy experiments were performed (Fig. 3b). The purified MWNTs show a typical Raman spectrum that contains a G band at 1580 cm−1 and a D band at 1342 cm−1. Upon oxidation, the intensity of the D band increased, and the G band shifted to a higher frequency (1583 cm−1), due to the introduction of oxygen-containing groups on the surface of the MWNTs. P3HT exhibits two Raman peaks at 1445 and 1378 cm−1, corresponding to the C–C skeletal stretching vibration and the CC skeletal stretching vibration, respectively.28 As expected, the Raman spectrum of the MWNT-g-P3HT includes feature peaks from both the MWNTs and P3HT. In addition, the G band of the MWNTs in the MWNT-g-P3HT is shifted to a lower frequency (1575 cm−1) compared to that of the purified and oxidized MWNTs (1580 and 1583 cm−1). This Raman feature reflects the electron transfer from the P3HT to the MWNTs in the MWNT-g-P3HT.28
The morphology of the purified MWNTs and the MWNT-g-P3HT was observed under TEM. Fig. 4a shows a TEM image of the purified MWNTs. It is seen that the MWNTs have a diameter of ca. 8 nm. A number of observations indicated that the purified MWNTs readily exist in an aggregated state. In contrast, the MWNT-g-P3HT existed as much smaller bundles, or individual tubes. This finding can be ascribed to the surface modification of the MWNTs: the grafted P3HT prevents intermolecular interactions between the MWNTs. Fig. 4b shows polymer bumps attached on the surface of an individual MWNT-g-P3HT. Since the un-grafted P3HT was removed by repeated washing during the purification, the polymer bumps must be covalently grafted on to the surface of the MWNTs. Moreover, the dispersibility of the MWNTs and the MWNT-g-P3HT was also confirmed by the stability of their suspensions in THF. It is seen that the MWNT-g-P3HT is readily dispersed in THF (inset in Fig. 4b), and that the purified MWNTs are likely to precipitate in THF (inset in Fig. 4a).
 | ||
| Fig. 4 TEM images of (a) the purified MWNTs, and (b) the MWNT-g-P3HT, with the insets showing the optical images of the purified MWNT suspension and the MWNT-g-P3HT suspension in THF. | ||
In order to investigate the impact of the MWNTs on the morphology and the crystalline structure of the MWNT-g-P3HT@P3HT composite films, TEM images and electron diffraction (ED) patterns were taken. Fig. 5a shows the TEM image of a P3HT film coated on an amorphous carbon film. It is seen that the P3HT forms isolated domains of ca. 500 nm in lateral size. This is due to the fact that the concentration of the P3HT solution used to prepare the film was relatively low, so the P3HT did not form a continuous film. On the isolated domains, one can also see the P3HT whisker crystals, which are the typical P3HT crystals. In contrast, with the influence of the MWNTs, the MWNT-g-P3HT@P3HT composite (MWNT-g-P3HT:P3HT = 1.5:1) readily formed a continuous film (Fig. 5b). Individual MWNTs or small bundles of MWNTs were uniformly dispersed in the composite film, while P3HT aggregates were likely to be found around the MWNTs. The influence of the MWNTs on the morphology development of the composite films is in agreement with our previous finding that the addition of single-walled carbon nanotubes (SWNTs) facilitates the formation of continuous composite films.32
 | ||
| Fig. 5 TEM images of (a) the P3HT film, and (b) the MWNT-g-P3HT@P3HT composite film (MWNT-g-P3HT:P3HT = 1.5:1) coated on amorphous carbon film; ED patterns of (a) the P3HT film, and (b) the MWNT-g-P3HT@P3HT composite film. | ||
Fig. 5c and d show the ED patterns of the P3HT film and the MWNT-g-P3HT@P3HT composite film. In order to study the influence of the MWNTs on the crystallinity of the MWNT-g-P3HT@P3HT composite film, both ED patterns were taken from films of the same thickness, using the same parameters, such as the selected-area aperture size and exposure time. Both ED patterns contain a dominant diffraction ring corresponding to the (020) plane of the P3HT crystals, where the π–π stacking of the P3HT backbones is located. Notably, the (020) diffraction ring in the ED pattern of the MWNT-g-P3HT@P3HT composite film is weaker than that of the P3HT film. This finding is contrary to the previous report that SWNTs enhance the crystallinity of the P3HT component in the composite film, due to the π–π stacking between the SWNTs and P3HT.32 The different response of the crystallinity of the P3HT to the CNTs might be due to the different interactions between the CNTs and the P3HT. Covalent grafting of P3HT chains on to the surface of the MWNTs must interfere with the π–π stacking between the P3HT and the surface of the MWNTs; as a result, the MWNTs interfere with the crystallization process of the P3HT in the MWNT-g-P3HT@P3HT composite film.
In order to evaluate the electrochemical sensitivity of the MWNT-g-P3HT@P3HT composite film for the detection of Hg2+ ions, Fig. 6a summarizes the CV curves of a GC electrode, and the P3HT film and the MWNT-g-P3HT@P3HT composite film (MWNT-g-P3HT:P3HT = 1.5:1) coated on to the tips of GC electrodes. In the CV curve for the GC electrode, an oxidation peak for Hg2+ ions was detected at 0.42 V. In contrast, the intensity of the oxidation peak increased and shifted to a higher potential (0.52 V) in the CV curve for the P3HT film. The increased electrochemical current of the oxidation peak is attributed to the preferential interaction between Hg2+ and the sulphur atoms in the P3HT main chains,33,34 whilst the shift of the oxidation peak to a higher potential indicates increased electrical resistance of the P3HT film compared to the GC electrode. Furthermore, upon incorporation of the MWNTs, the MWNT-g-P3HT@P3HT composite film yields a further enhanced electrochemical current of the oxidation peak; meanwhile, the oxidation peak shifts to a lower potential (0.48 V) compared to that of the P3HT film. The decreased potential of the oxidation peak indicates that the electrical conductivity of the composite film is higher than that of the P3HT film, due to the incorporation of MWNTs in the composite film. On the other hand, the increased intensity of the oxidation peak for the MWNT-g-P3HT@P3HT composite film electrode is ascribed to the synergistic effect of the electrocatalytic properties from the MWNTs and P3HT.
 | ||
| Fig. 6 (a) CV curves of a GC electrode, the P3HT film coated on to the tip of a GC electrode, and the MWNT-g-P3HT@P3HT composite film (MWNT-g-P3HT:P3HT = 1.5:1) coated on to the tip of a GC electrode in the presence of Hg2+ ions (Hg(NO3)2·H2O, 5.88 μM in aqueous solution); (b) current maxima of the oxidation peaks obtained with MWNT-g-P3HT@P3HT composite films having different ratios of MWNT-g-P3HT to P3HT. | ||
Fig. 6b summarizes the influence of the ratio of MWNT-g-P3HT to P3HT in the MWNT-g-P3HT@P3HT composite film on the electrochemical sensitivity for the detection of Hg2+ ions in an aqueous solution (5.88 μM). The composite films prepared with a component ratio of 1:1 and 1.5:1 show the highest electrochemical currents of the oxidation peak, amongst the several ratios tested. This result can be attributed to the balance between the sensitivities of P3HT and the MWNTs to Hg2+ ions, so the synergistic effect of the MWNTs and P3HT is most thoroughly exhibited at such ratios. At a higher mass ratio of MWNT-g-P3HT to P3HT, i.e. 2.5:1, the composite shows reduced electrocatalytic ability, which is even worse than that of P3HT. This result might be ascribed to the phase separation between the MWNTs and P3HT, as evidenced by TEM observation (not shown).
Fig. 7 summarizes the quantitative detection of Hg2+ ions using the MWNT-g-P3HT@P3HT composite film (MWNT-g-P3HT:P3HT = 1.5:1) electrode. In the CV curves (Fig. 7a), it is seen that the intensities of the oxidation peak and the reduction peak increase with increased concentration of Hg2+ ions, from 0 to 5.88 μM, with an increasing step of 0.588 μM. The lowest detection limit is 0.588 μM, i.e. 0.2 mg L−1 for Hg(NO3)2·H2O. Fig. 7b shows the change in the electrochemical current of the oxidation peak as a function of the concentration of Hg2+ ions. This result indicates that the MWNT-g-P3HT@P3HT composite film electrode can be used for the quantitative electrochemical detection of Hg2+ ions.
 | ||
| Fig. 7 (a) CV curves for the MWNT-g-P3HT@P3HT composite film (MWNT-g-P3HT:P3HT = 1.5:1) electrode in the presence of various concentrations of Hg2+ ions (from 0 to 5.88 μM with an increasing step of 0.588 μM); (b) the change in the electrochemical current of the oxidation peak as a function of the concentration of Hg2+ ions. | ||
Conclusions
In this research, we modified MWNTs by grafting P3HT on to their surfaces through an amidation reaction between the carboxylic acid groups on the MWNTs, and the amine groups of an amino-terminated P3HT. Such surface modification increased the dispersibility of the MWNTs in the P3HT matrix, leading to enhanced capability of the MWNT-g-P3HT@P3HT composite to form continuous films, and improved electrical properties of the composite films. Importantly, the MWNT-g-P3HT@P3HT composite films showed improved electrochemical sensitivity for the quantitative detection of Hg2+ ions.Acknowledgements
This work was supported by the “Hundred Talents Program” of the Chinese Academy of Sciences, the National Natural Science Foundation of China (21274158, 91333114, and 20902077), the Key Laboratory of Oil & Gas Fine Chemicals, Ministry of Education & Xinjiang Uyghur Autonomous Region, Xinjiang University (XJDX0908-2011-04), and the start-up foundation for doctors in Xinjiang University (BS080118).Notes and references
- S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923–925 CrossRef CAS.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- R. Po, M. Maggini and N. Camaioni, J. Phys. Chem. C, 2010, 114, 695–706 CAS.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- Z. Yao, Y. Yang, X. Chen, X. Hu, L. Zhang, L. Liu, Y. Zhao and H.-C. Wu, Anal. Chem., 2013, 85, 5650–5653 CrossRef CAS PubMed.
- Z. Yao, B. Huang, X. Hu, L. Zhang, D. Li, M. Guo, X. Zhang, H. Yuan and H.-C. Wu, Analyst, 2013, 138, 1649–1652 RSC.
- C. Guo, X. Yang, X. Wang, M. Pei and G. Zhang, New J. Chem., 2013, 37, 4163–4169 RSC.
- V. C. Goncalves and D. T. Balogh, Sens. Actuators, B, 2012, 162, 307–312 CrossRef CAS PubMed.
- M. Laurenti, E. Lopez-Cabarcos, F. Garcia-Blanco, B. Frick and J. Rubio-Retama, Langmuir, 2009, 25, 9579–9584 CrossRef CAS PubMed.
- Y. Lattach, F. Garnier and S. Remita, ChemPhysChem, 2012, 13, 281–290 CrossRef CAS PubMed.
- V. Dua, S. P. Surwade, S. Ammu, X. Zhang, S. Jain and S. K. Manohar, Macromolecules, 2009, 42, 5414–5415 CrossRef CAS.
- P. H. Lepage, R. Peytavi, M. G. Bergeron and M. Leclerc, Anal. Chem., 2011, 83, 8086–8092 CrossRef PubMed.
- M. Liu, C. Luo and H. Peng, Talanta, 2012, 88, 216–221 CrossRef CAS PubMed.
- H. Peng, L. Zhang, C. Soeller and J. Travas-Sejdic, Biomaterials, 2009, 30, 2132–2148 CrossRef CAS PubMed.
- B. Fang, S. Jiao, M. Li, Y. Qua and X. Jiang, Biosens. Bioelectron., 2008, 23, 1175–1179 CrossRef CAS PubMed.
- L. Zhang, H. Sun, D. Li, S. Song, C. Fan and S. Wang, Macromol. Rapid Commun., 2008, 29, 1489–1494 CrossRef CAS.
- A. Uygun, Talanta, 2009, 79, 194–198 CrossRef CAS PubMed.
- H. Guan, P. Zhou, X. Zhou and Z. He, Talanta, 2008, 77, 319–324 CrossRef CAS PubMed.
- M. Liu, B. Li and X. Cui, Talanta, 2013, 115, 837–841 CrossRef CAS PubMed.
- Z. Yao, H. Bai, C. Li and G. Shi, Chem. Commun., 2011, 47, 7431–7433 RSC.
- C. Li, M. Numata, M. Takeuchi and S. Shinkai, Angew. Chem., Int. Ed., 2005, 44, 6371–6374 CrossRef CAS PubMed.
- R. Zhan, Z. Fang and B. Liu, Anal. Chem., 2010, 82, 1326–1333 CrossRef CAS PubMed.
- M.-P. Plante, E. Berube, L. Bissonnette, M. G. Bergeron and M. Leclerc, ACS Appl. Mater. Interfaces, 2013, 5, 4544–4548 CAS.
- X. Chen, T. Ren, M. Ma, Z. Wang, G. Zhan and C. Li, Electrochim. Acta, 2013, 111, 49–56 CrossRef CAS PubMed.
- C.-Z. Li, H. Karadeniz, E. Canavar and A. Erdem, Electrochim. Acta, 2012, 82, 137–142 CrossRef CAS PubMed.
- L. Aguei, C. Pena-Farfal, P. Yanez-Sedeno and J. M. Pingarron, Electrochim. Acta, 2007, 52, 7946–7952 CrossRef CAS PubMed.
- M. Jeffries-El, G. Sauve and R. D. McCullough, Macromolecules, 2005, 38, 10346–10352 CrossRef CAS.
- D. Meng, J. Sun, S. Jiang, Y. Zeng, Y. Li, S. Yan, J. Geng and Y. Huang, J. Mater. Chem., 2012, 22, 21583–21591 RSC.
- J. Sun, G. Wu and J. Geng, Polym. J., 2013, 45, 813–818 CrossRef CAS.
- J. X. Geng, B. S. Kong, S. B. Yang, S. C. Youn, S. Park, T. Joo and H. T. Jung, Adv. Funct. Mater., 2008, 18, 2659–2665 CrossRef CAS.
- B. K. Kuila, K. Park and L. Dai, Macromolecules, 2010, 43, 6699–6705 CrossRef CAS.
- J. X. Geng and T. Y. Zeng, J. Am. Chem. Soc., 2006, 128, 16827–16833 CrossRef CAS PubMed.
- H. Zejli, J. L. H.-H. de Cisneros, I. Naranjo-Rodriguez and K. R. Temsamani, Talanta, 2007, 71, 1594–1598 CrossRef CAS PubMed.
- A. Jimenez-Morales, J. C. Galvan and P. Aranda, Electrochim. Acta, 2002, 47, 2281–2287 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |