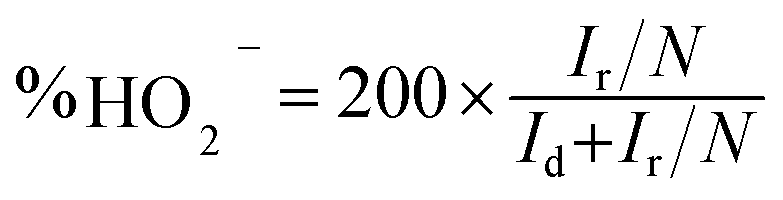DOI:
10.1039/C4RA02208D
(Paper)
RSC Adv., 2014,
4, 26653-26661
Electrocatalytic activity of metalloporphyrins grown in situ on graphene sheets toward oxygen reduction reaction in an alkaline medium†
Received
13th March 2014
, Accepted 28th May 2014
First published on 28th May 2014
Abstract
A series of non-noble-metal catalysts for oxygen reduction reaction (ORR), based on metal 5,10,15,20-tetrakis(4-hydroxyphenyl)porphyrin (M-THPP, M: Fe3+, Co2+, Ni2+, Mn2+) grown on poly(sodium-p-styrenesulfonate) modified reduced graphene oxide (PSS-rGO), were fabricated using an in situ solvothermal synthesis method. The morphology of the M-THPP/PSS-rGO was characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Ultraviolet-visible (UV-vis) absorption spectroscopy and X-ray photoelectron spectroscopy (XPS) techniques were utilized to analyse the unusual interactions between the metalloporphyrins and graphene sheets. Electrochemical measurements using rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE) techniques were employed to study the catalytic activity and the mechanism of the oxygen reduction reaction on the as-synthesised M-THPP/PSS-rGO catalysts in an alkaline medium. The half-wave potential for the ORR on the CoTHPP/PSS-rGO catalyst was found to be around −0.22 V vs. SCE, which was much higher than those on the other M-THPP/PSS-rGO catalysts and similar to that on Pt/C (−0.20 V vs. SCE). RDE and RRDE results show that the ORR process proceeds mainly via an almost 4-electron pathway on CoTHPP/PSS-rGO. The catalyst stability tests disclose that the CoTHPP/PSS-rGO is much more stable than the other M-THPP/PSS-rGO composites. The assembled CoTHPP/PSS-rGO catalyst possesses high activity, good long-term stability, excellent tolerance to the crossover effect of methanol and a facile 4-electron pathway for ORR, which could be used as a promising Pt-free catalyst in an alkaline direct methanol fuel cell.
1. Introduction
With its high power density, high energy-conversion efficiency, scalability and low to zero emission of pollutants, direct methanol fuel cells (DMFC) have received considerable attention during the past few decades.1–3 DMFCs consist of a Pt–Ru anode for methanol oxidation, a Pt cathode for oxygen reduction, and a proton exchange membrane (PEM).1 DMFC operates by oxidizing methanol to CO2 and reducing oxygen to water.1,3,4 Usually, the electrochemical reduction of O2 is a multi-electron reaction with slow kinetics.5,6 In alkaline media, the ORR mechanism can be expressed using reactions (1) and (2),| | |
O2 + H2O + 2e− → HO2− + OH−
| (1) |
| | |
O2 + 2H2O + 4e− → 4OH−
| (2) |
To the best of our knowledge, the transfer of two electrons to produce H2O2 (HO2−) is potentially hazardous and less exoergic.6,7 To achieve maximum energy-efficiency capacity, it is highly desirable to reduce O2 via a direct four-electron pathway. In this respect, Pt-based materials or Pt is known to be the most efficient catalyst since it provides the highest current response towards the direct four-electron reduction of oxygen to water.6,8–12 Due to not only the rarity and expensiveness but also the poor tolerance to the crossover effect and instability of Pt,6,13–15 numerous non-precious catalysts, including heteroatom-(boron, nitrogen, sulfur, phosphorus) doped carbon nanomaterials,16–21 metal oxide/carbide/nitride materials,22–26 transition-metal chalcogenides,27,28 N-containing polymers29–32 and MN4-centers of transition metal N4-macrocycles,33–39 have been developed and screened in terms of their oxygen reduction reaction (ORR) for replacing Pt. Among them, transition metal N4-macrocycles, especially transition metal porphyrins and phthalocyanines, have received remarkable attention, since Jasinski used cobalt phthalocyanine as an electrocatalyst for ORR,43 due to their large π-conjugated aromatic systems with unique structures and coordination properties,40,41 thermal and chemical robustness,40 and rich electronic properties.42 Therefore, a numerous transition metal N4-macrocycles with different substitutes and central metals have been designed and applied to fabricate modified electrodes for the ORR.44–46 However, as semiconductor materials,40 all these transition metal N4-macrocycles still suffer from low activity and stability in the ORR, and are thus far from satisfying the requirements for the commercialization of fuel cells.
Carbon nano-materials (CNMs), such as carbon powder, graphene, carbon nanotubes, are more suitable catalyst supports due to their interesting physicochemical properties47–49 such as excellent electrical conductivity, large specific surface area and high chemical stability.40 Recently, several researchers have concentrated on CNMs-supported transition metal N4-macrocycles materials. CNMs are used to support the transition metal N4-macrocycles because strong π–π interactions between the CNMs and transition metal N4-macrocycles can make transition metal N4-macrocycle moieties closely load on CNMs, and thus stabilizing the system. In addition, the most important aspect is that the formation of micro/nano composites could provide more active sites and create synergistic effect, which significantly improves the catalytic activity for the ORR.47,49 Mamuru et al.50 used octabutylsulphonylphthalocyanine complexes of iron (FeOBSPc) and cobalt (CoOBSPc) supported on multi-walled carbon nanotube (MWCNT) platforms for the electrocatalytic reduction of oxygen, and found that the MWCNT–FeOBSPc composite exhibited the best ORR activity involving a direct 4-electron mechanism. Jahan et al.51 investigated the electrocatalytic properties of a graphene–metalloporphyrin composite, which showed a facile 4-electron ORR pathway with significantly higher selectivity towards ORR and significant tolerance to the crossover effect of methanol when compared with the Pt/C catalyst. Morozan and co-workers52 investigated cobalt and iron phthalocyanines or porphyrins supported on different carbon nanotubes towards the oxygen reduction reaction. Mo et al.53 synthesized a novel FeTSPc/SWCNTs composite by a facile modification of single-walled carbon nanotubes (SWCNTs) with iron tetrasulfophthalocyanine (FeTSPc), and the hybrid material displayed better specificity, long-term stability, tolerance to methanol crossover effect and resistance towards CO poisoning effect than that of the commercial Pt/C catalyst.
To date, CNMs-supported transition metal N4-macrocycle complexes, as electrocatalytic materials, are generally fabricated using the following methods: (1) π–π interactions;51,54 (2) layer-by-layer (LBL) assembly;55 (3) solid-phase synthesis;40 (4) covalent modification;56 and (5) solvothermal synthesis.57,58 Among these methods, solvothermal synthesis is considered to be an effective and commercially viable method due to its advantages such as the feasibility of controlling the morphology, accessibility of cheap raw materials, low growth temperature and relatively economic processing. Our group57,58 have prepared iron phthalocyanine/graphene and cobalt tetranitrophthalocyanine/graphene micro/nano composites for the ORR through solvothermal processes. These two catalysts exhibited high catalytic activity, an almost 4-electron ORR pathway, much higher long-term stability and tolerance to methanol crossover effect compared with that of the Pt/C catalyst. In this study, a series of non-noble-metal catalysts for the ORR, including FeTHPP/PSS-rGO, CoTHPP/PSS-rGO, NiTHPP/PSS-rGO and MnTHPP/PSS-rGO, were fabricated by an in situ solvothermal synthesis method similar to our previous report.58 The electrocatalytic performance and ORR mechanism of the M-THPP/PSS-rGO composites were discussed based on cyclic voltammetry, rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE) measurements. Among the fabricated electrocatalysts, it was found that the CoTHPP/PSS-rGO composite displayed high activity, good long-term stability, excellent tolerance to the crossover effect of methanol and an almost direct 4-electron pathway toward ORR in an alkaline solution, which could be viewed as a promising cathode candidate for alkaline direct methanol fuel cell applications.
2. Experimental section
2.1 Chemicals
5,10,15,20-Tetrakis(4-hydroxyphenyl)porphyrin (THPP) was synthesised as described in the literature.59 Pyrrole and p-hydroxy benzaldehyde were obtained by Aladdin. Natural graphite powder was purchased from Sigma-Aldrich. Reagents including H2SO4 (98%), NaNO3, KMnO4, H2O2 (30%), NaOH, metal chloride, DMF, propionic acid were of analytical grade and were used without further purification. Poly(sodium-p-styrenesulfonate) (PSS), hydrazine hydrate and Pt/C (20 wt% Pt on Vulcan XC-72) were purchased from Alfa Aesar. Distilled water was used throughout the experiments.
2.2 Synthesis of graphite oxide (GO) and poly(sodium-p-styrenesulfonate) modified reduced graphene oxide (PSS-rGO)
GO was synthesised from graphite powder by a modified Hummers' method.60,61 PSS-rGO was obtained according to a reported procedure.62 First, a GO aqueous colloidal suspension (200 mL, 1 mg mL−1) was prepared by ultrasonically dispersing the paper-like graphite oxide in distilled water. Then, 2 g of poly(sodium-p-styrenesulfonate) was added with constant stirring for 12 h at ambient temperature and pressure. Afterwards, 4 mL of hydrazine hydrate was slowly added to the mixture, which was heated to 95 °C and continuously stirred for 24 h. After cooling to room temperature, the mixture was separated by centrifugation and washed five times with distilled water. Finally, the precipitate was dispersed into DMF by ultrasonication, resulting in the PSS-rGO suspension (1.4 mg mL−1).
2.3 Synthesis of M-THPP/PSS-rGO composites
The M-THPP/PSS-rGO composites were prepared by an in situ solvothermal synthesis method. The synthesis routes to obtain M-THPP/PSS-rGO are shown in Scheme 1. In a typical experiment, THPP (0.1031 mmol, 7 mg), metal chloride (0.3093 mmol) and PSS-rGO (5 mL, 1.4 mg mL−1) dispersion were added into 10 mL of DMF. The mixture was then sonicated for 30 min. Subsequently, the stable suspension was sealed in a Telfon-lined autoclave and solvothermally treated at 150 °C for 6 h, and then cooled to room temperature. The composite suspension was centrifuged, and the residue was sequentially washed three times with DMF, distilled water and alcohol.
 |
| | Scheme 1 The synthesis routes to M-THPP/PSS-rGO composites. | |
2.4 Characterization
For morphological characterization, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images were collected on a JSM-6701F field emission scanning electron microscope operating at 5 kV and JEOL JEM-1200EX transmission electron microscope operating at 100 kV, respectively. UV-vis spectra were recorded using a mini UV-1240 spectrophotometer. The surface characterization of the synthesized samples was conducted by X-ray photoelectron spectroscopy (XPS, ESCLAB 250 spectrometer) using an Al Kα X-ray source (1486.6 eV photons).
2.5 Electrode preparation and electrochemical measurements
A catalyst ink (1.0 mg mL−1) was prepared by ultrasonically blending 10 mg of the catalyst with 10 mL of alcohol. The ink was deposited onto a freshly polished glassy carbon (GC) electrode, and the coated electrode was then left to air dry. The total catalyst loading on the GC electrode was calculated to be 0.283 mg cm−2. For comparison, the Pt/C or PSS-rGO modified GC electrode was also prepared using the same procedures described above.
Electrochemical measurements, including cyclic voltammetry (CV), rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE), were accomplished by a computer-controlled potentiostat (CHI660E electrochemical workstation, CH Instrument, USA) with a three-electrode cell system, in which the modified GC electrode with a catalyst was employed as the working electrode, a platinum-wire electrode as the auxiliary electrode, and a saturated calomel electrode (SCE) as the reference electrode. Cyclic voltammetry measurements were conducted at the GC electrode (CHI104, 3 mm in diameter) in a N2- or O2-saturated 0.1 M NaOH electrolyte. The potential range was cyclically scanned between −0.8 V and +0.4 V vs. SCE at a scan rate of 100 mV s−1 at room temperature. RDE measurements were conducted at the GC rotating-disk electrode (5 mm in diameter) in an O2-saturated 0.1 M NaOH solution with a scan rate of 10 mV s−1. Linear sweep voltammetry measurements (LSV) were scanned between −0.8 V and +0.2 V vs. SCE at different rotating speeds from 100 to 2500 rpm. Long-term chronoamperometric experiments were performed at a constant rotation speed of 1600 rpm. The RRDE data were obtained using a Pine Instrument Company AF-MSRCE modulator speed rotator. The working electrode was a glassy carbon disk (5.61 mm in diameter) and a platinum ring (collection efficiency N = 0.37). An O2-saturated 0.1 M NaOH aqueous solution was used as the electrolyte. The potential of the disk electrode was varied from −0.8 V to +0.2 V vs. SCE at a scan rate of 10 mV s−1, and the ring potential was maintained at 0.1 V vs. SCE.
3. Results and discussion
3.1 Characterization of M-THPP/PSS-rGO composites
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to observe the surface morphology of the prepared samples. As shown in Fig. 1A and C, the PSS-rGO displays a flexibly aggregated, crumpled nanosheet structure. For CoTHPP/PSS-rGO (Fig. 1B and D), we can observe nanoparticles closely stacked on the graphene sheet surface. The average diameter of the nanoparticles is around 25 nm. Fig. 1E shows the TEM image of only CoTHPP nanoparticles fabricated via an in situ solvothermal synthesis method. The morphology and average diameter of CoTHPP nanoparticles is the same as the nanoparticles coated on graphene, demonstrating that the nanoparticles on the surface of graphene correspond to cobaltoporphyrin.
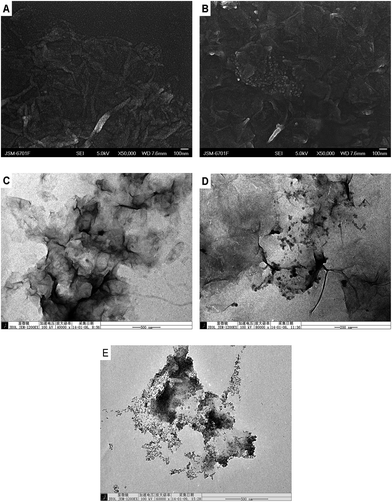 |
| | Fig. 1 SEM images of (A) PSS-rGO and (B) CoTHPP/PSS-rGO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), at higher (×50000) magnification; TEM images of (C) PSS-rGO, (D) CoTHPP/PSS-rGO (1:1) and (E) CoTHPP, at higher magnification (×40000, ×80000 and ×60000, respectively). :1), at higher (×50000) magnification; TEM images of (C) PSS-rGO, (D) CoTHPP/PSS-rGO (1:1) and (E) CoTHPP, at higher magnification (×40000, ×80000 and ×60000, respectively). | |
The formation of the M-THPP/PSS-rGO hybrid material should be related to the following facts. The negatively charged PSS modified on graphene can not only prevent the graphene sheets from stacking, but can also adsorb the positively charged metal ions by electrostatic interactions. Meanwhile, the adsorbed metal ions react with metal-free porphyrin molecules at solvothermal conditions, and M-THPP is synthesized and coated in situ on to the PSS-rGO. The M-THPP molecules adsorbed on the graphene can serve as “nuclei” for the π–π assembly between the M-THPP moieties and PSS-rGO sheets, leading to the formation of a randomly aggregated micro/nano hybrid.
The unusual interactions between the metalloporphyrins and PSS-rGO sheets were monitored by a UV-vis spectrometer and X-ray photoelectron spectrometer (XPS), respectively. Fig. 2a shows the UV-vis spectra of PSS-rGO, CoTHPP and CoTHPP/PSS-rGO in an alcohol solution. It can be seen that the bare PSS-rGO shows a distinct absorption peak at 267 nm, and the CoTHPP displays a strong Soret band peak at 432 nm and two weaker Q band peaks at 543 nm and 584 nm, respectively. However, the CoTHPP/PSS-rGO composite exhibits a Soret band absorbance (436 nm) with a red-shift of 4 nm compared with CoTHPP. Furthermore, the Soret band absorbance of other M-THPP/PSS-rGO composites also displays different degrees of red-shift (ESI, Fig. S1†). These results indicate that there are π–π interactions between the M-THPP moieties and PSS-rGO sheets.
 |
| | Fig. 2 (a) UV-vis absorption spectra of PSS-rGO, CoTHPP and CoTHPP/PSS-rGO; (b) XPS spectra of PSS-rGO and CoTHPP/PSS-rGO. | |
In addition to using UV-vis spectra, the interactions between the metalloporphyrin moieties and PSS-rGO sheets were confirmed by XPS. By comparison of the XPS survey spectra of PSS-rGO and M-THPP/PSS-rGO, we can confirm the presence of metalloporphyrins in M-THPP/PSS-rGO as characteristic N 1s peaks exist in all the composites prepared (shown in Fig. 2b and ESI, Fig. S2†). In addition to the N 1s signals, various metal 2p signals in Fig. 2b and Fig. S2† are also visible due to the metallic center of porphyrin. Fig. 3a and b show the N 1s high-resolution XPS spectra for CoTHPP/PSS-rGO and CoTHPP. From the decomposition of the N 1s XPS spectrum, the spectrum for cobaltoporphyrin can be well fitted to two peaks with a roughly equal area. The higher binding energy signal corresponds to the pyrrolic-like nitrogen, and the lower binding energy signal to the pyridine-like nitrogen.63,64 In contrast to CoTHPP, for CoTHPP/PSS-rGO, the N 1s peaks of the pyrrolic- and pyridine-like nitrogen atoms are shifted to higher binding energies by 0.16 eV and 0.44 eV, respectively (Fig. 3a and b). From Fig. 3c and d, it can be seen that, compared with free CoTHPP, the binding energies of Co 2p in CoTHPP/PSS-rGO are shifted from 781.29 eV (2p3/2) and 796.38 eV (2p1/2)65 to 781.76 eV (2p3/2) and 797.62 eV (2p1/2), respectively. For other M-THPP/PSS-rGO composites, the same phenomenon observed with CoTHPP/PSS-rGO can also be seen (ESI, Fig. S3†). Such a pronounced shift in either N 1s or metal 2p in binding energy further demonstrates that strong electronic interactions exist between the metalloporphyrin moieties and PSS-rGO sheets.
 |
| | Fig. 3 High-resolution N 1s and Co 2p XPS spectra of (a), (c) CoTHPP/PSS-rGO and (b), (d) CoTHPP, respectively. | |
3.2 Electrochemical behavior of M-THPP/PSS-rGO in the oxygen reduction reaction
Fig. 4a shows the cyclic voltammetry curves of the CoTHPP/PSS-rGO and THPP/PSS-rGO modified electrodes in either an N2 or O2 saturated 0.1 M NaOH solution. As shown in Fig. 4a, in the presence of N2 saturation, no distinguished current response can be observed on THPP/PSS-rGO when the potential was cycled between −0.8 and 0.40 V vs. SCE. In contrast, the cyclic voltammetry curve of the CoTHPP/PSS-rGO presents a pair of nearly symmetrical redox peaks (0.1 V vs. SCE), which should be attributed to the redox transformation of the CoN4 center. In Fig. 4a, however, a well-defined oxygen reduction peak (−0.24 V vs. SCE) can be observed on the CoTHPP/PSS-rGO composite when cyclic voltammetry was conducted in an O2-saturated 0.1 M NaOH solution, suggesting the high catalytic activity of the CoTHPP/PSS-rGO composite towards the ORR.
 |
| | Fig. 4 Cyclic voltammetry curves of (a) CoTHPP/PSS-rGO and THPP/PSS-rGO in either an N2 or O2 saturated 0.1 M NaOH solution. (b) CoTHPP/PSS-rGO, CoTHPP and PSS-rGO in either an N2 or O2 saturated 0.1 M NaOH. Scan rate: 100 mV s−1. | |
ESI, Fig. S4† displays the cyclic voltammogram curves of CoTHPP/PSS-rGO with different mass ratios of THPP to PSS-rGO in an O2-staturated 0.1 M NaOH solution. In Fig. S4,† when the initial ratio of THPP to PSS-rGO was 2:1, the peak potential (Ep) and peak current density of the ORR on CoTHPP/PSS-rGO can be observed at around −0.34 V vs. SCE and 1.98 mA cm−2, respectively. When the mass ratio of THPP to PSS-rGO varies from 2:1 to 1:1, the cathodic peak potential for oxygen reduction gradually shifts to a more positive value from −0.34 V to −0.24 V vs. SCE, and the peak current density also increases to 3.32 mA cm−2. Whereas, when the mass ratio of THPP to PSS-rGO varies from 1:1 to 1:6, the peak current density increases slowly but the Ep shifts to a more negative value. Therefore, the CoTHPP/PSS-rGO composite with a 1:1 mass ratio of THPP to PSS-rGO was employed in the following study because of the most positive Ep value and higher peak current density.
Fig. 4b depicts the cyclic voltammetry curves of the CoTHPP/PSS-rGO, PSS-rGO and CoTHPP modified GC electrodes in either an N2 or O2 saturated 0.1 M NaOH solution. The ORR Ep value of CoTHPP/PSS-rGO is 103 mV and 197 mV vs. SCE more positive than those of PSS-rGO and CoTHPP, respectively, and the current density on CoTHPP/PSS-rGO is more than twice as large as those on PSS-rGO and CoTHPP. These results strongly indicate that the catalytic activity of CoTHPP was significantly improved when graphene was used as a support, suggesting the synergistic effect of CoTHPP and PSS-rGO towards the ORR.
3.3 Mechanism of the oxygen reduction reaction on the M-THPP/PSS-rGO composites
To quantitatively verify the ORR mechanism, our materials were loaded onto GC electrodes for rotating-disk electrode (RDE) measurements. Fig. 5a shows the RDE curves for CoTHPP/PSS-rGO at various rotation speeds (from 100 to 2500 rpm). The limiting current depicts a typical increase with rotation speeds due to a shortened diffusion layer.16 Similar studies for other catalysts, such as FeTHPP/PSS-rGO, MnTHPP/PSS-rGO, NiTHPP/PSS-rGO and PSS-rGO, are provided in the supplementary information (Fig. S5(a1–4)).† Fig. 5b shows a series of the RDE curves of different catalysts including commercial Pt/C (20 wt%) measured in O2-saturated 0.1 M NaOH at a scan rate of 10 mV s−1 and rotation speed of 1600 rpm. To compare the activity of different catalysts with each other, the half-wave potential of each catalyst was estimated by determining the maxima of the derivatives of the reduction current (see Table 1 for details). Note that the CoTHPP/PSS-rGO is the most active catalyst among fabricated catalysts due to its high half-wave potential (−0.22 V vs. SCE), followed by MnTHPP/PSS-rGO (−0.32 V vs. SCE), FeTHPP/PSS-rGO (−0.38 V vs. SCE) and NiTHPP/PSS-rGO (−0.40 V vs. SCE); however, it does not outperform the commercial Pt/C catalyst (−0.20 V vs. SCE). The electrocatalytic activity of these hybrids appears to be considerably affected by the nature of the central metal. In addition, we also compared our catalyst with some recently reported similar hybrid materials using graphene and MN4 complexes that have admirable performance for cathodic ORR (see ESI, Table S1†). In general, the performance of our CoTHPP/PSS-rGO catalyst is comparable with, or even better than previously reported catalysts in terms of the onset potential, half-wave potential and limiting current density.
 |
| | Fig. 5 (a) Linear sweep voltammograms of CoTHPP/PSS-rGO at different rotating speeds; (b) linear sweep voltammograms of PSS-rGO, M-THPP/PSS-rGO and Pt/C at a rotation speed of 1600 rpm. (c) Koutecky–Levich plots of CoTHPP/PSS-rGO and (d) electron transfer number of PSS-rGO and M-THPP/PSS-rGO at fixed potentials of −0.4 V, −0.5 V, −0.6 V and −0.7 V vs. SCE. | |
Table 1 Electrochemical parameters for oxygen reduction estimated from the polarization curves at 1600 rpm
| Catalyst |
E1/2/V vs. SCE |
JL/mA cm−2 |
Tafel plot slopes (mV dec−1) |
| Low η |
High η |
| PSS-rGO |
−0.34 |
3.51 |
139 |
538 |
| NiTHPP/PSS-rGO |
−0.40 |
5.16 |
117 |
484 |
| MnTHPP/PSS-rGO |
−0.32 |
5.11 |
113 |
449 |
| FeTHPP/PSS-rGO |
−0.38 |
6.15 |
83 |
454 |
| CoTHPP/PSS-rGO |
−0.22 |
5.16 |
61 |
366 |
| Pt/C |
−0.20 |
5.33 |
61 |
227 |
To shed light on the electrochemical kinetics of the catalysts in the ORR, Tafel analysis was performed on these samples. Fig. S6† presents the Tafel curves, which were derived from the polarization curves of Fig. 5b with a rotation rate of 1600 rpm. It can be seen that different catalysts lead to different Tafel slope values. The Tafel slopes on various samples are listed in Table 1. Clearly, the Tafel slope on CoTHPP/PSS-rGO catalyst gives the smallest value in the low or high overpotential region. In general, in the low overpotential region, the first electron transfer is the rate-determining step, and the oxygen reduction process is controlled by the kinetics of the surface reaction.67 In the high overpotential region, the reaction rate is dominated by the diffusion limitation inside the material, which is commonly interpreted in terms of a two-electron transfer reaction as the rate-determining step.57
On the basis of the RDE voltammograms, we can calculate the number of electrons transferred (n) according to the Koutecky–Levich equation:33,37,51,68
| |
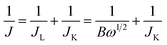 | (3) |
| | |
B = 0.62nFC0D02/3ν−1/6
| (4) |
where
J is the measured current density (mA cm
−2),
JK and
JL are the kinetic and diffusion-limited current densities, respectively,
n is the electron transfer number for the reaction and
F is the Faraday constant (96
500 C mol
−1).
D0 is the diffusion coefficient for oxygen in solution (2.0 × 10
−5 cm
2 s
−1),
ν is the kinematic viscosity of the electrolyte solution (0.001 cm
2 s
−1),
C0 is the concentration of O
2 in the air-saturated solution (0.25 mM), and
ω is the rotation speed (rpm s
−1). All the parameters are invariant over the potential range with the exception of the
n value. By plotting the graph between
J−1 and
ω−1/2, the
n value can be determined based on the slope of Koutecky–Levich plots.
Fig. 5c displays the Koutecky–Levich plots of CoTHPP/PSS-rGO for ORR at fixed potentials of −0.4 V, −0.5 V, −0.6 V and −0.7 V vs. SCE, respectively, which were derived from the RDE curves of CoTHPP/PSS-rGO obtained at various rotation speeds. Similar Koutecky–Levich plots for other catalysts are given in the ESI, Fig. S5(b1–4).† According to the Koutecky–Levich equation, the calculated n values for the ORR on M-THPP/PSS-rGO are shown in Fig. 5d. However, Fig. 5d shows that the average electron transfer number n on most of the M-THPP/PSS-rGO composites presents an even bigger number than 4 over the entire potential range. The reason, according to previous study,53,66 might be that the oxygen reduction pathways occurring on our materials were very complicated, involving not only the ORR but also the redox process of the porphyrin ring. This implies that the Koutecky–Levich equation was not reliable to accurately evaluate the electron transfer number per O2 molecule.
To accurately confirm the ORR mechanism of the M-THPP/PSS-rGO composites, a rotating ring disk electrode (RRDE) technique was employed. The electron number (n) and hydrogen peroxide ion yield (% HO2−) during ORR can be determined by the following equations:46,54,69
| |
 | (5) |
| |
 | (6) |
where
N = 0.37 is the collection efficiency,
Id is the disk current, and
Ir is the ring current.
Fig. 6a shows the Id and Ir values for FeTHPP/PSS-rGO, CoTHPP/PSS-rGO, NiTHPP/PSS-rGO and MnTHPP/PSS-rGO. The corresponding Ir value for the oxidation of hydrogen peroxide ion (HO2−) was measured with a Pt ring electrode at a polarized potential of 0.10 V vs. SCE. On the basis of Id and Ir values, the electron transfer number (n) was calculated to be 3.69–3.80 for FeTHPP/PSS-rGO, 3.79–3.83 for CoTHPP/PSS-rGO, 3.19–3.72 for MnTHPP/PSS-rGO and 2.65–2.82 for NTHPP/PSS-rGO over the potential range of −0.8 V to +0.20 V vs. SCE (Fig. 6b). Fig. 6c displays the percentage of hydrogen peroxide ion yielded on the MTHPP/PSS-rGO composites in the same potential range. The trends in Fig. 6b and c indicate that the ORR mechanism is strongly potential-dependent. Note that the CoTHPP/PSS-rGO composite with n = 3.79–3.83 and % HO2− = 8.60–10.70%, exhibits much higher activity over the entire potential range, emphasizing that the ORR process on CoTHPP/PSS-rGO proceeds mainly via a direct four-electron pathway.
 |
| | Fig. 6 (a) RRDE tests for the ORR on M-THPP/PSS-rGO in O2-saturated 0.1 M NaOH at a rotation speed of 1600 rpm. The ring electrode is polarized at 0.1 V vs. SCE. Scan rate: 10 mV s−1. (b) Electron transfer number and (c) hydrogen peroxide ion percentage of CoTHPP/PSS-rGO, FeTHPP/PSS-rGO, MnTHPP/PSS-rGO and NiTHPP/PSS-rGO as a function of electrode potential. | |
3.4 Study of stability and methanol crossover effect
The long-term stability of catalysts is one of the major concerns in current DMFC technology. The stability of M-THPP/PSS-rGO and Pt/C was assessed through chronoamperometric measurements for 12 h in an O2-saturated 0.1 M NaOH aqueous solution at a rotation rate of 1600 rpm. As revealed in Fig. 7, the chronoamperometric response for CoTHPP/PSS-rGO exhibits a very slow attenuation with the highest current retention of 75%. In contrast, the corresponding current loss for the Pt/C catalyst shows a fast decrease with the lowest retention (46%) under the same conditions. Fig. 8 shows the cyclic voltammetry curves of CoTHPP/PSS-rGO (a) and Pt/C (b) in an O2-saturated 0.1 M NaOH aqueous solution, in the presence/absence of 3 M methanol. In the absence of methanol, the obvious ORR peaks at −0.24 V and −0.21 V vs. SCE can be observed for CoTHPP/PSS-rGO and Pt/C, respectively. After adding 3 M methanol, for Pt/C, the ORR peak current decreases significantly and a new oxidation peak at 0.0 V vs. SCE appears (Fig. 8b), while no notable change in both peak potential and peak current is observed for CoTHPP/PSS-rGO (Fig. 8a). Thus, we believe that our CoTHPP/PSS-rGO catalyst possesses excellent stability and can eliminate the negative effects of the methanol crossover.
 |
| | Fig. 7 The chronoamperometry curves of Pt/C and M-THPP/PSS-rGO in O2-saturated 0.1 M NaOH with a rotation speed of 1600 rpm. | |
 |
| | Fig. 8 Cyclic voltammetry curves of CoTHPP/PSS-rGO (a) and Pt/C (b) at a scan rate of 100 mV s−1 in O2-saturated 0.1 M NaOH solution with and without 3 M CH3OH. | |
4. Conclusions
In summary, we have fabricated novel M-THPP/PSS-rGO composites via an in situ solvothermal synthesis method. SEM and TEM results demonstrated that the CoTHPP moieties were tightly anchored on to the PSS-rGO surface. UV-vis and XPS spectra results suggested that there were strong π–π interactions between the M-THPP moieties and PSS-rGO sheets. Electrochemical measurements results disclosed that among the fabricated electrocatalysts, CoTHPP/PSS-rGO possessed the highest electrocatalytic activity, the best long-term stability, excellent tolerance to the crossover effect of methanol, especially, the more efficient 4-electron pathway towards the ORR in an alkaline solution, which could be anticipated for mass-production as the cathode catalyst candidate for alkaline direct methanol fuel cells.
Acknowledgements
The work was supported by the Natural Science Foundation of China (no. 21273024) and the Natural Science Foundation of Jilin Province, China (no. 201215135).
References
- S. Sundarrajan, S. I. Allakhverdiev and S. Ramakrishna, Int. J. Hydrogen Energy, 2012, 37, 8765–8786 CrossRef CAS PubMed
 .
. - X. Zhao, M. Yin, L. Ma, L. Liang, C. P. Liu, J. H. Liao, T. H. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 CAS .
- Y. H. Lee, G. Lee, J. H. Shim, S. Hwang, J. Kwak, K. Lee, H. Song and J. T. Park, Chem. Mater., 2006, 18, 4209–4211 CrossRef CAS .
- Y. F. Zhang, Z. Y. Yuan, S. B. Wang, H. He, Y. R. Zhao and X. W. Liu, Energy Fuels, 2010, 24, 6449–6455 CrossRef CAS .
- Z. Q. Jiang, Z. J. Jiang, X. N. Tian and W. H. Chen, J. Mater. Chem. A, 2014, 2, 441–450 CAS .
- H. J. Yin, H. J. Tang, D. Wang, Y. Gao and Z. Y. Tang, ACS Nano, 2012, 6, 8288–8297 CrossRef CAS PubMed .
- A. Trojánek, J. Langmaier, S. Záliš and Z. Samec, Electrochim. Acta, 2013, 110, 816–821 CrossRef PubMed .
- K. Sasaki, H. Naohara, Y. M. Choi, Y. Cai, W. F. Chen, P. Liu and R. R. Adzic, Nat. Commun., 2012, 3, 1115–1124 CrossRef PubMed .
- D. L. Wang, Y. C. Yu, H. L. Xin, R. Hovden, P. Ercius, J. A. Mundy, H. Chen, J. H. Richard, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nano Lett., 2012, 12, 5230–5238 CrossRef CAS PubMed .
- N. R. Elezović, B. M. Babić, Lj. Gajić-Krstajić, P. Ercius, V. R. Radmilović, N. V. Krstajić and Lj. M. Vračar, Electrochim. Acta, 2012, 69, 239–246 CrossRef PubMed .
- Z. W. Chen, M. Waje, W. Z. Li and Y. S. Yan, Angew. Chem., Int. Ed., 2007, 46, 4060–4063 CrossRef CAS PubMed .
- R. Lin, H. Y. Zhang, T. T. Zhao, C. H. Cao, D. J. Yang and J. X. Ma, Electrochim. Acta, 2012, 62, 263–268 CrossRef CAS PubMed .
- C. W. B. Bezerra, L. Zhang, K. C. Lee, H. S. Liu, A. L. B. Marques, E. P. Marques, H. J. Wang and J. J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS PubMed .
- C. X. Zhang, K. Yanagisawa, H. J. Tao, A. Onda, T. Shou and S. Kamiya, Catal. Lett., 2012, 142, 1128–1133 CrossRef CAS .
- Y. W. Ma, H. M. Zhang, H. X. Zhong, T. Xu, H. Jin, Y. F. Tang and Z. Xu, Electrochim. Acta, 2010, 55, 7945–7950 CrossRef CAS PubMed .
- W. Yang, T. P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed .
- D. H. Deng, X. L. Pan, L. Yu, Y. Cui, Y. P. Jiang, J. Qi, W. X. Li, Q. Fu, X. C. Ma, Q. K. Xue, G. Q. Sun and X. H. Bao, Chem. Mater., 2011, 23, 1188–1193 CrossRef CAS .
- D. S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed .
- R. Silva, D. A. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 25, 7823–7826 CrossRef PubMed .
- W. Y. Wonga, W. R. W. Daud, A. B. Mohamad, A. A. H. Kadhum, K. S. Loh and E. H. Majlan, Int. J. Hydrogen Energy, 2013, 38, 9370–9386 CrossRef PubMed .
- S. B. Yang, L. J. Zhi, K. Tang, X. L. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS .
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed .
- Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed .
- I. Roche, E. Chaînet, M. Chatenet and J. Vondrák, J. Phys. Chem. C, 2007, 111, 1434–1443 CAS .
- Y. Ohgi, A. Ishihara, K. Matsuzawa, S. Mitsushima, K. I. Ota, M. Matsumoto and H. Ima, Electrochim. Acta, 2012, 68, 192–197 CrossRef CAS PubMed .
- J. H. Kim, A. Ishihara, S. Mitsushima, N. Kamiya and K. I. Ota, Electrochim. Acta, 2007, 52, 2492–2497 CrossRef CAS PubMed .
- Y. J. Feng and N. Alonso-Vante, Phys. Status Solidi B, 2008, 245, 1792–1806 CrossRef CAS .
- M. R. Gao, Y. F. Xu, J. Jiang and S. H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC .
- S. W. Yuan, J. L. Shui, L. Grabstanowicz, C. Chen, S. Commet, B. Reprogle, T. Xu, L. P. Yu and D. J. Liu, Angew. Chem., 2013, 125, 8507–8511 CrossRef .
- S. H. Lim, Z. T. Li, C. K. Poh, L. F. Lai and J. Y. Lin, J. Power Sources, 2012, 214, 15–21 CrossRef CAS PubMed .
- J. Wu, W. M. Li, D. Higgins and Z. W. Chen, J. Phys. Chem. C, 2011, 115, 18856–18862 CAS .
- Q. Zhou, C. M. Li, J. Li and J. T. Lu, J. Phys. Chem. C, 2008, 112, 18578–18583 CAS .
- U. B. Nasini, Y. Gartia, P. Ramidi, A. Kazi, A. U. Shaikh and A. Ghosh, Chem. Phys. Lett., 2013, 566, 38–43 CrossRef CAS PubMed .
- W. M. Li, A. P. Yu, D. C. Higgins, B. G. Llanos and Z. W. Chen, J. Am. Chem. Soc., 2010, 132, 17056–17058 CrossRef CAS PubMed .
- M. Isaacs, M. J. Aguirre, A. Toro-Labbé, J. Costamagna, M. Páez and J. H. Zagal, Electrochim. Acta, 1998, 43, 1821–1827 CrossRef CAS .
- B. Wang, J. Power Sources, 2005, 152, 1–15 CrossRef CAS PubMed .
- I. Kruusenberg, J. Mondal, L. Matisen, V. Sammelselg and K. Tammeveski, Electrochem. Commun., 2013, 33, 18–22 CrossRef CAS PubMed .
- S. R. Sun, N. Jiang and D. G. Xia, J. Phys. Chem. C, 2011, 115, 9511–9517 CAS .
- R. L. Liu, C. V. Malotki, L. Arnold, N. Koshino, H. Higashimura, M. Baumgarten and K. Müllen, J. Am. Chem. Soc., 2011, 133, 10372–10375 CrossRef CAS PubMed .
- H. J. Li, Z. W. Xu, K. Z. Li, X. H. Hou, G. X. Cao, Q. L. Zhang and Z. Y. Cao, J. Mater. Chem., 2011, 21, 1181–1186 RSC .
- X. Feng, L. Chen, Y. P. Dong and D. L. Jiang, Chem. Commun., 2011, 47, 1979–1981 RSC .
- H. He, Y. K. Lei, C. Xiao, D. Y. Chu, R. R. Chen and G. F. Wang, J. Phys. Chem. C, 2012, 116, 16038–16046 CAS .
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS .
- W. Chen, J. Akhigbe, C. Brückner, C. M. Li and Y. Lei, J. Phys. Chem. C, 2010, 114, 8633–8638 CAS .
- S. Swavey and A. Eder, Inorg. Chem. Commun., 2013, 29, 14–17 CrossRef CAS PubMed .
- J. Masa and W. Schuhmann, Chem.–Eur. J., 2013, 19, 9644–9654 CrossRef CAS PubMed .
- N. C. Cheng, C. Kemna, S. Goubert-Renaudin and A. Wieckowski, Electrocatalysis, 2012, 3, 238–251 CrossRef CAS PubMed .
- W. Zhang, A. U. Shaikh, E. Y. Tsui and T. M. Swager, Chem. Mater., 2009, 21, 3234–3241 CrossRef CAS .
- G. L. Turdean, I. C. Popescu, A. Curulli and G. Palleschi, Electrochim. Acta, 2006, 51, 6435–6441 CrossRef CAS PubMed .
- S. A. Mamuru, K. I. Ozoemena, T. Fukuda, N. Kobayashi and T. Nyokong, Electrochim. Acta, 2010, 55, 6367–6375 CrossRef CAS PubMed .
- M. Jahan, Q. L. Bao and K. P. Loh, J. Am. Chem. Soc., 2012, 134, 6707–6713 CrossRef CAS PubMed .
- A. Morozan, S. Campidelli, A. Filoramo, B. Jousselme and S. Palacin, Carbon, 2011, 49, 4839–4847 CrossRef CAS PubMed .
- G. Q. Mo, S. Y. Liao, Y. Z. Zhang, W. D. Zhang and J. S. Ye, Electrochim. Acta, 2012, 76, 430–439 CrossRef CAS PubMed .
- Y. Y. Jiang, Y. Z. Lu, X. Y. Lv, D. X. Han, Q. X. Zhang, L. Niu and W. Chen, ACS Catal., 2013, 3, 1263–1271 CrossRef CAS .
- H. J. Tang, H. J. Yin, J. Y. Wang, N. L. Yang, D. Wang and Z. Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 1–6 CrossRef .
- S. K. Kim and S. Jeon, Electrochem. Commun., 2012, 22, 141–144 CrossRef CAS PubMed .
- L. L. Cui, G. J. Lv, Z. Y. Dou and X. Q. He, Electrochim. Acta, 2013, 106, 272–278 CrossRef CAS PubMed .
- G. J. Lv, L. L. Cui, Y. Y. Wu, Y. Liu, T. Pu and X. Q. He, Phys. Chem. Chem. Phys., 2013, 15, 13093–13100 RSC .
- A. C. Serra, M. Pineiro, A. M. d'A. Rocha Gonsalves, M. Abrantes, M. Laranjo, A. C. Santos and M. F. Botelho, J. Photochem. Photobiol., B, 2008, 92, 61–67 CrossRef PubMed .
- Y. X. Xu, L. Zhao, H. Bai, W. J. Hong, C. Li and G. Q. Shi, J. Am. Chem. Soc., 2009, 131, 13490–13497 CrossRef CAS PubMed .
- J. X. Geng and H. T. Jung, J. Phys. Chem. C, 2010, 114, 8227–8234 CAS .
- D. Q. Wu, F. Zhang, H. W. Liang and X. L. Feng, Chem. Soc. Rev., 2012, 41, 6160–6177 RSC .
- A. Gulino, Anal. Bioanal. Chem., 2013, 405, 1479–1495 CrossRef CAS PubMed .
- D. M. Sarno, L. J. Matienzo and W. E. Jones, J. Inorg. Biochem., 2001, 40, 6308–6315 CrossRef CAS PubMed .
- K. Artyushkova, S. Levendosky, P. Atanassov and J. Fulghum, Top. Catal., 2007, 46, 263–275 CrossRef CAS .
- Y. Yuan, J. Ahmed and S. Kim, J. Power Sources, 2011, 196, 1103–1106 CrossRef CAS PubMed .
- M. T. de Groot, M. Merkx, A. H. Wonders and M. T. M. Koper, J. Am. Chem. Soc., 2005, 127, 7579–7586 CrossRef CAS PubMed .
- R. R. Chen, H. X. Li, D. Y. Chu and G. F. Wang, J. Phys. Chem. C, 2009, 113, 20689–20697 CAS .
- T. S. Olson, S. Pylypenko, J. E. Fulghum and P. Atanassov, J. Electrochem. Soc., 2010, 157, B54–B63 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02208d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.