Heavy atom tunneling in the automerization of pentalene and other antiaromatic systems†
Sebastian Kozuch*
Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, 1155 Union Circle #305070, Denton, Texas 76203-5070, USA. E-mail: seb.kozuch@gmail.com
First published on 5th May 2014
Abstract
Cyclobutadiene is a well-known system that can automerize (i.e. undergo a π bond-shifting) by a heavy atom tunneling mechanism. To understand the rules that allow this process, a theoretical study has been carried out on the contribution of tunneling to the automerization reactions of several other molecules with antiaromatic π systems: pentalene, heptalene, acepentalene, and substituted pentalenes. The calculations find that automerization of molecules such as pentalene, which have planar structures, are most likely to proceed by rapid carbon tunneling from the lowest vibrational state, since such molecules have relatively low activation energy and narrow barriers. However, if a molecule is not planar (thus formally “non-aromatic”) and/or requires large geometry changes in order to reach the automerization transition state, then the tunneling will be strongly hindered. In some cases, such as heptalene and tri-tert-butylpentalene, the rearrangement of the reactant requires a modest amount of thermal energy, which can be followed by the π bond-shifting through a tunneling mechanism (“thermally activated tunneling”).
Introduction
Shifting of the π bonds in cyclobutadiene (CBD) was the first reaction in which, through a combination of experiments and calculations, tunneling by carbon was shown to be important. The anomalously low pre-exponential factor for the automerization of cyclobutadiene-d2 (eqn (1)),
 | (1) |
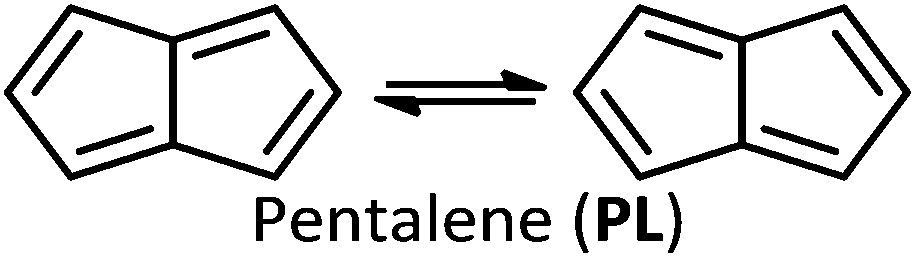
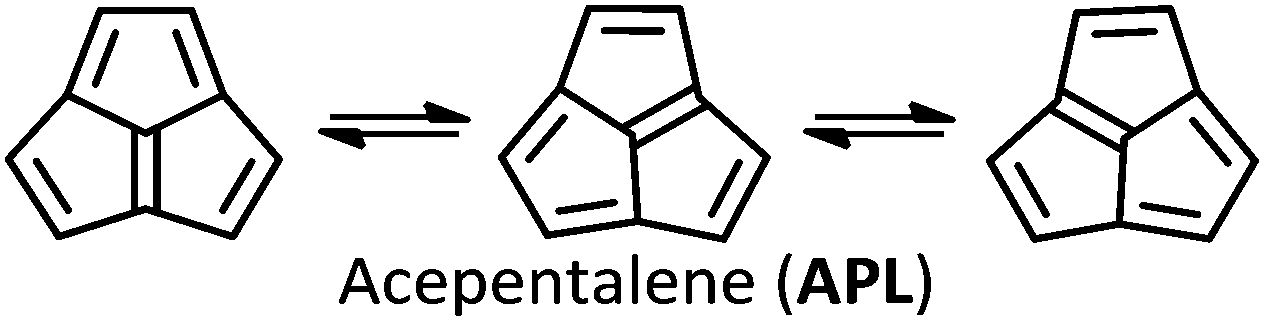
Like CBD, pentalene (PL), acepentalene (APL), and heptalene (HL) are antiaromatic, containing alternant π bonds.‡ As shown in eqn (2)–(4), PL, HL and APL can, like CBD, undergo automerization by shifting of the π bonds.
 | (2) |
 | (3) |
 | (4) |
All three of these molecules have alternating bond lengths, caused by pseudo-Jahn–Teller distortions of the most symmetrical geometries.15–17 They are highly reactive, with a tendency to dimerize even at low temperature.18–21
HL was first synthesized in 1961,22 and the elusives APL and PL in 1995 and 1996,17,18 respectively (although sterically shielded or electronically stabilized derivatives have been known for some decades19–21,23–25). In all these cases the dianion is a stable aromatic compound,17,26 with the capacity to act as an organometallic ligand.27
In this paper the results of a computational investigation of the extent to which QMT is involved in the automerization reactions of eqn (2)–(4) (including some substituted systems) are reported. In addition, an analysis of the experimental results in the automerizations of 1,3,5-tri-tert-butyl-pentalene24 (tBu3PL) and of HL28 is tackled, with both reactions occurring faster than the NMR time-scale at low temperatures.29
Computational methodology
Degeneracy of the Frontier orbitals
Although there are many similarities between CBD on one hand and PL, HL and APL on the other, there is one obvious difference – PL, HL and APL each contain C–C bonds across the antiaromatic annulene rings. The existence of these bonds make their electronic structures different from that of CBD in one important way. At the geometries of highest symmetry, CBD contain a pair of half-filled MOs that are degenerate by symmetry. In contrast, in PL, HL and APL the cross-ring bond between the bridgehead carbons lifts this degeneracy by allowing the 2p AOs on these carbons to interact,18 thus creating non-alternant systems.30 As shown in Fig. 1, this interaction does not affect the LUMO of PL or APL, but it stabilizes the HOMO. On the other hand, in HL, it is the LUMO that is destabilized by this interaction and the HOMO that is unaffected. | ||
| Fig. 1 HOMO and LUMO of PL, HL, and APL, and SOMOs of CBD in their highest symmetry, corresponding to the transition state of the automerization reaction (symmetries D4h, D2h, D2 and Cs, respectively). | ||
The degeneracy of the pair of singly occupied molecular orbitals (SOMOs) in square CBD means that a two determinantal electronic wave function must be used at and near the square geometry (the TS).31 In contrast, provided that the HOMO-LUMO gap is sufficiently large in the other molecules, a single-configuration wave function should suffice. This difference should make calculations of the automerization rates much simpler for PL, HL and APL (eqn (2)–(4)) than for CBD (eqn (1)).
Rate constant calculations
Rate constants for passage over the reaction barriers were computed using canonical variational transition state theory (CVT)32 and the contributions of multi-dimensional tunneling were incorporated using the small curvature tunneling (SCT) approximation33 with step size of 0.001 Bohr and quantized reactant state tunneling (QRST) for the reaction coordinate mode. This approach has proven to be successful in previous theoretical studies.13,14 The rate constants were computed with Polyrate,34 using Gaussrate35 as the interface between Polyrate and Gaussian 09.36 Unless specified, SCT values include the CVT rate constant and the tunneling correction.SCT calculations require calculation of energies, energy gradients, and second derivatives, not only at the reactant and TS geometries but also at many points along the reaction pathway. High-quality, wave-function-based calculations are not practical for SCT. Therefore, a search for a DFT functional that would provide high-quality results with a small basis set was carried out. As a benchmark against which DFT results with different functionals could be judged, the relative energy between the reactant and TS (ΔE‡) for the automerization of PL and HL was computed at the CCSD(T)-F12/cc-pVTZ-F12//M06-2X/6-311G(d) level of theory. Molpro37,38 was used to perform these calculations. Values of ΔE‡ = 11.3 and 14.9 kcal mol−1 were obtained, respectively, for PL and HL.39 The values of the T1 diagnostics were 0.017 for both transition states. In addition, an (8/8)CASSCF calculation on PL found that the weight of the second most important configuration, in which the LUMO is doubly occupied, relative to the configuration in which the HOMO is doubly occupied, is only 0.09/0.75 = 0.12. This indicates that single-reference CCSD(T) calculations are adequate for these reactions.
The ΔE‡ values for twenty DFT functionals were computed and compared to the CCSD(T)-F12/cc-pVTZ-F12 benchmark. The 6-31G(d) basis set was used for these DFT calculations, which were all performed with Gaussian 09.36 The values obtained from these calculations are given in the ESI.† The values that were closest to the CCSD(T)-F12/cc-pVTZ-F12 ones were obtained with M06-2X.40 Therefore, this functional was selected for all the QMT calculations presented here (see the ESI† for a discussion on the accuracy of unrestricted M06-2X for CBD).
The tBu3PL molecule is too large to allow calculation of its SCT rate constant. In order to simulate the effect of the mass of the tert-butyl groups in tBu3PL,24 we also calculated the rate constants for automerization of 1,3,5-tribromopentalene, Br3PL, (whose bromine substituents have a mass almost four times higher than the tert-butyls of tBu3PL), and with a model system of PL with the hydrogens in the first, third and fifth position having a mass of 57 (the mass of a tBu group and without the electronic effects of Br atoms). However, the tBu groups may rotate during the automerization reaction, while the atomic Br and 57H cannot mimic this movement. This may be a significant factor in the tunneling regime, hence we also calculated the automerization of 1,3,5-trimethylpentalene (Me3PL) and an hypothetical trimethylpentalene with the hydrogens of the methyls having a mass of 15 (the mass of CH3), to model the tBu substituent rotation (15Me3PL).
Results
Table 1 and Fig. 2 present the rate constants with and without tunneling corrections (CVT in blue and SCT in red). Classical rate constants give linear Arrhenius plots, consistent with the Arrhenius formulation (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k = lnA − Ea/RT). However, QMT processes are temperature independent (at least when the reaction occurs from the ground vibrational state), and therefore a plateau in the Arrhenius plot indicates a tunnelling mechanism.
k = lnA − Ea/RT). However, QMT processes are temperature independent (at least when the reaction occurs from the ground vibrational state), and therefore a plateau in the Arrhenius plot indicates a tunnelling mechanism.
| PL | Br3PL | 57H3PL | Me3PL | 15Me3PL | HL | APLa | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔE‡ | 11.3 | 9.0 | 11.3 | 11.9 | 11.9 | 13.0 | 7.3 | |||||||
| T (K) | CVT | SCT | CVT | SCT | CVT | SCT | CVT | SCT | CVT | SCT | CVT | SCT | CVT | SCT |
| a Since APL has three equivalent minima, the rate constants should be doubled to account for the two possible automerizations. | ||||||||||||||
| 10 | 2.1 × 10−196 | 2.2 × 108 | 1.4 × 10−145 | 2.8 × 108 | 4.8 × 10−196 | 8.8 × 107 | 9.6 × 10−197 | 7.2 × 10−1 | 1.9 × 10−204 | 1.5 × 10−25 | 1.6 × 10−240 | 1.2 × 10−38 | 1.3 × 10−126 | 1.9 × 10−9 |
| 50 | 4.2 × 10−30 | 2.3 × 108 | 6.5 × 10−20 | 3.9 × 108 | 5.2 × 10−30 | 1.1 × 108 | 4.4 × 10−30 | 3.2 × 102 | 9.2 × 10−32 | 4.0 × 10−1 | 7.0 × 10−39 | 1.4 × 10−10 | 3.6 × 10−16 | 5.0 × 10−8 |
| 100 | 4.7 × 10−9 | 3.3 × 108 | 5.7 × 10−4 | 7.9 × 108 | 5.1 × 10−9 | 1.9 × 108 | 5.2 × 10−9 | 1.8 × 105 | 2.9 × 10−10 | 5.3 × 103 | 2.1 × 10−13 | 3.6 × 10−1 | 3.8 × 10−2 | 3.4 × 10−1 |
| 150 | 5.9 × 10−2 | 5.2 × 108 | 1.4 × 102 | 1.6 × 109 | 6.2 × 10−2 | 3.5 × 108 | 5.6 × 10−2 | 2.9 × 106 | 3.6 × 10−3 | 1.6 × 105 | 7.9 × 10−5 | 1.2 × 103 | 2.1 × 103 | 4.9 × 103 |
| 200 | 2.3 × 102 | 8.6 × 108 | 7.8 × 104 | 3.0 × 109 | 2.4 × 102 | 6.5 × 108 | 1.7 × 102 | 1.6 × 107 | 1.2 × 101 | 1.1 × 106 | 1.7 × 100 | 9.5 × 104 | 5.1 × 105 | 8.4 × 105 |
| 300 | 1.0 × 106 | 2.5 × 109 | 4.7 × 107 | 1.0 × 1010 | 1.0 × 106 | 2.2 × 109 | 4.9 × 105 | 1.4 × 108 | 3.5 × 104 | 1.1 × 107 | 3.8 × 104 | 1.2 × 107 | 1.3 × 108 | 1.7 × 108 |
| 400 | 7.0 × 107 | 7.6 × 109 | 1.2 × 109 | 3.0 × 1010 | 7.0 × 107 | 7.0 × 109 | 2.4 × 107 | 6.3 × 108 | 1.7 × 106 | 5.0 × 107 | 6.0 × 106 | 2.0 × 108 | 2.1 × 109 | 2.4 × 109 |
 | ||
| Fig. 2 Arrhenius graphs for the automerization reaction of PL, HL, APL, Br3PL, Me3PL, 15Me3PL and 57H3PL. In blue the CVT (classical) rate values. In red the SCT (QMT) values. | ||
At 10 K the tunnelling calculations predict very fast bond-shifting in PL, Br3PL and 57H3PL, with extremely short half-lives of 3 × 10−9, 2 × 10−9 and 8 × 10−9 s, respectively (computed as ln(2)/k). According to these calculations, these systems can shift their π bonds at such a pace that it will be virtually impossible to observe the alternating bond lengths by common experimental procedures (e.g., NMR) at any temperature. Therefore, for all practical purposes, pentalene and two of the substituted pentalenes will not show alternating bonds and will appear to have a single symmetrical geometry, in spite of the fact that they actually consist of two equivalent structures in fast equilibrium (i.e., they each have a “double well” potential energy surface).
In the case of the rate constants for automerization of Me3PL a plateau is also reached, but only at a much lower temperature (approximately at 20 K) with a much longer half time (t1/2 = 1 s) in this temperature regime.
15Me3PL, HL and APL do not reach such a plateau except at extremely low temperatures (below 20 K, see ESI†). However, the SCT rate constants for bond shifting in 15Me3PL and HL still have values that are much larger than the CVT ones, which do not include tunneling (for APL this only occurs below 100 K). This difference can be attributed to thermally activated tunneling (caused by large atom displacements, as described below), where the occupation of vibrationally excited states results in a lower and narrower effective barrier.
Discussion
Pentalene (PL) and derivatives
PL has a flat Arrhenius profile, indicating very fast QMT from the ground state for the bond-shifting reaction at a broad range of temperatures (see Table 1 and Fig. 2). This is an indication that heavy atom tunneling may be possible for the degenerate rearrangement of antiaromatic molecules other than CBD. Even at high temperatures automerization of PL is predicted to proceed by tunnelling, albeit from vibrationally excited states. At room temperature, the SCT rate constant of PL automerization is still computed to be three orders of magnitude larger than the CVT rate constant.Fig. 3 shows the energy of the system vs. the displacement of the carbon atoms with the wider trajectory. It is possible to see that the barrier profile for PL is very narrow, an optimal factor for a fast QMT (see Fig. 4 for the difference between single and double bond distances of these molecules).
 | ||
| Fig. 3 Energy vs. displacement curves for the highlighted carbon atoms. The TS is set at zero displacement. Note that since multidimensional tunneling can “cut corners”, to proceed through the least action pathway,41 the actual trajectories may differ slightly from the one-dimensional pathways in this figure. | ||
 | ||
| Fig. 4 C–C bond distances in Å at the optimized geometries of the three anti-aromatic molecules. | ||
The rate of automerization in PL has not been measured experimentally, but low-temperature (−50 °C) NMR experiments found that the peaks in the 13C NMR spectra of 1,3,5-tri-tert-butylpentalene (tBu3PL) showed no indication of bond localization.24 Unfortunately, tBu3PL has too much conformational flexibility and too many atoms to make SCT calculations on its automerization practical.
Therefore, in order to investigate the effect of the masses of the three tert-butyl groups on the rate of automerization, calculations on simpler models were performed, including 1,3,5-tribromo-pentalene (Br3PL), and a pentalene with three, hypothetical, super-heavy, 57H isotopes (57H3PL) at C1, C3, and C5, in order to mimic the masses of the tBu groups that are attached to these carbons in tBu3PL. The SCT rate constants (Table 1 and Fig. 2) are very similar to those for the automerization of the unsubstituted PL (the ΔE‡ for automerization of Br3PL is lower than that for PL, but not enough to affect the kinetics). These results predict that the masses of the substituents in these PL derivatives will not have much effect on the rates of tunnelling. The reason for this lack of sensitivity to the substituent mass is that these substituents are almost immobile throughout the automerization reaction, and therefore their masses are not determining factors for QMT.
However, tBu3PL has several different conformers that are produced by the rotation of the three tert-butyl substituents, with the energies of these conformers affected by the positions of the double bonds in the eight-membered PL ring. As will be discussed shortly, this means that the tert-butyl groups must rotate as the double bonds in tBu3PL shift. This effect cannot be mimicked by calculations on Br3PL or 57H3PL.
Therefore, a study of 1,3,5-trimethylpentalene (Me3PL) was carried out, in order to model the effect of the tert-butyl group conformations. Of course, the effective masses of the hydrogens of a rotating methyl group in Me3PL are very different from those of a rotating tert-butyl group in tBu3PL, and this difference in effective mass could make the probability of tunneling very different in these molecules.13 Therefore, we also performed QMT calculations on 15Me3PL, an hypothetical molecule with an isotopic mass of 15 on the hydrogens of the methyl groups, since the mass of 15H is equivalent to the mass of the methyl groups of each tert-butyl substituent. This model is far from perfect, because the C–H bonds in the methyl groups are ca. 0.4 Å shorter than the C–C bonds in the tert-butyl groups. However, calculations on 15Me3PL should at least provide an upper limit to the rate of automerization by tunneling of tBu3PL.
The preferred conformation of a methyl substituent is one in which one C–H bond eclipses the C–C π bond of the doubly bonded carbons.42 In the reactant of Me3PL all three methyl groups have this conformation. However, as the methyl groups in the product also have this preferred conformation, they must rotate as the double bonds shift, as depicted in Fig. 5. Thus, for the automerization of Me3PL to be completely degenerate, the bond-shifting and rotation of the three methyl groups by 60° must be coupled.
 | ||
| Fig. 5 Automerization of Me3PL, including the rotation of the methyl groups. At the TS there is a rotation of 60° of the left-upper Me group, and 30° of the lower one. | ||
The eclipsed conformation between a C–H and an adjacent π bond is favored by ∼2 kcal mol−1 over the staggered conformation.42 However, the overall ΔE‡ for Me3PL automerization is only 0.6 kcal mol−1 higher than that of PL (see Table 1), much less than the sum of three Me rotations (∼6 kcal mol−1) and an independent PL bond-shifting (11.3 kcal mol−1). This occurs because both reactions (π bond-shifting and methyl rotations) are, in fact, coupled, one aiding the other.
The Arrhenius plots for automerization of Me3PL and 15Me3PL are substantially different from that of PL at low temperatures (Table 1 and Fig. 2). At 10 K the automerization of Me3PL is predicted to be fast, with t1/2 = 1 s (but still more than eight orders of magnitude slower than that of PL).
In contrast to Me3PL, the rate of automerization of 15Me3PL at 10 K is computed to be essentially zero. To the extent that the hypothetical 15H3C groups model the tert-butyl groups, calculations predict that tBu3PL will not automerize at cryogenic temperatures.
As shown by the results of our calculations on Br3PL and 57H3PL, the masses of the substituents do not affect the tunneling rates for the bond-shifting. Since the automerization barrier heights are very similar for all the PL based systems, the disparity between the Arrhenius plots for automerization of Me3PL, 15Me3PL, and PL must be attributed to the rotation of the Me groups. The fifteen times greater mass of the hydrogens in the hypothetical 15Me3PL, compared to the actual masses of hydrogens in Me3PL produces a computed tunneling rate at 10 K 1024 times slower.
The low barriers to methyl group torsions make a relatively easy thermal crossing of this barrier. Consequently, on-going from 10 K to 100 K, although the rate of automerization of unsubstituted PL is calculated to increase by only a factor of 1.5, the increase for 15Me3PL is calculated to be greater than a factor of 1028, due to a “thermally activated tunneling” process.
What do computational results predict for the rate of automerization of tBu3PL? Its behaviour will resemble the model 15Me3PL system: at cryogenic temperatures it would be impossible to have rotation of the bulky tBu groups, and therefore the automerization will be virtually impossible. However, the tert-butyl rotation requires almost the same energy as the methyl groups of Me3PL (ca. 2 kcal mol−1 per substituent). With a calculated ΔE‡ similar to all the other PL derivatives for the degenerate rearrangement (12.3 kcal mol−1), it will undergo a thermally activated tunneling at higher temperatures as was experimentally observed,24 showing an apparently delocalized system instead of the localized “Kekule” structures (see the ESI† for the XYZ structures and a description of the conformers of tBu3PL).
Heptalene (HL) and acepentalene (APL)
Considering the substantial impact of QMT on PL, it may be expected that this effect can be generalized to other anti-aromatic species. Therefore, an analysis of heptalene (eqn (3)) and of acepentalene (eqn (4)) was carried out.In NMR studies, HL showed a gradual broadening below −120 °C and later splitting of the 13C signals at −160 °C,28 an indication of a fast alternation of π bonds from two localized structures. At that temperature range, the activation energy for the process was estimated to be 3.5 kcal mol−1, a fact that baffled the authors28 of the study, since the calculated barrier was of 12 kcal mol−1 (not far from the 13.0 kcal mol−1 calculated here). From the calculations, we can now explain this paradox as the outcome of a tunneling process: an estimation of the SCT “Arrhenius style” activation energy (Ea = −R∂lnk/∂T−1) gives a value of 5 kcal mol−1 at the same experimental temperatures, comparable to the experimental results.
If the tunneling effect is so evident in HL, why does it not start from the ground vibrational state (i.e. reaching a plateau in the Arrhenius plot, see Fig. 2)? Similar to tBu3PL, HL must rearrange its geometry prior to the π bond-shifting. The optimized HL is a puckered D2 non-planar geometry.‡ However, the π bond-shifting requires an almost planar geometry, as shown in Fig. 6. As a result, the molecule undergoes a “flapping” movement, inverting the concavity after the π bond-shifting, as can be seen in Fig. 6. This flapping motion has a large amplitude, especially for the outermost carbon atoms.
 | ||
| Fig. 6 Side view of the deformation of heptalene and acepentalene in the bond-shifting reaction. | ||
In Fig. 3 the displacement graph of this outermost carbon of HL (in purple) shows these two orthogonal movements. At the beginning, there is a broad and flat curve, corresponding to the planarization (the first half of the flapping). Such a wide trajectory is difficult to overcome by a tunneling mechanism, but its low energy profile is easy to “climb” by a small amount of thermal energy. It is possible to calculate the flapping inversion without π bond-shifting, providing a ΔE‡ of 4.7 kcal mol−1 (not surprisingly close to the “Arrhenius style” activation energy); at mild temperatures this barrier can be easily surmounted.
Once the (almost) planar geometry is reached, the sharp energy peak of the π bond-shifting is ideal for a QMT crossing, but not for a classical “over the barrier” mechanism. As a result, the SCT calculation shows a much faster reaction than CVT (7 orders of magnitude at 150 K); still, at low enough temperatures (without a sufficient amount of thermal energy to flatten the molecule) tunneling through the automerization barrier will be impossible.
At −160 °C the calculated t1/2 for the automerization is 0.14 s, and at −120 °C 4 × 10−4 s. These results are compatible with the experimental NMR observations of broadening of the 13C signal at the latter and splitting at the former temperature,28 taking into account the NMR time scales.§29 The matching experimental/theoretical results supports the results of the SCT calculations, and corroborates the QMT hypothesis of the automerization of HL.
As for APL, it shows a negligible tunneling correction in spite of the fact that the single/double bond length difference (Fig. 4) is not much different to PL. However, APL has a bowl-shaped asymmetric geometry, which must be restructured to its specular structure, as seen if Fig. 6. This process involves a wide movement of atoms, broadening the displacement curve (green curve in Fig. 3). Opposed to HL, in APL the deformation of the bowl and the bond-shifting are intertwined in a smooth, wide reaction profile, not an ideal situation for QMT.
We can deduce from these results that automerization of antiaromatic molecules can proceed by QMT only when the system is planar (as in CBD and PL), or can be easily rearranged to a planar system by infusing a moderate amount of thermal energy (as in tBu3PL and HL).
Conclusions
A study of automerization reactions by heavy atom quantum mechanical tunneling was carried out on several antiaromatic molecules. In a nutshell, in some of these systems the π bond-shifting has the ideal conditions to proceed through carbon tunneling from the ground state (relatively low ΔE‡, but most critically very narrow barriers), as can be seen in pentalene. However, if the system is not planar and/or requires rearranging of the molecular geometry, then the tunneling will be strongly hindered.Indeed, the comparison of heptalene and acepentalene on one side, and pentalene on the other, shows that the former molecules, being non-planar, do not undergo facile QMT, as they require a large displacement of atoms to achieve a “kosher” planar geometry for the bond-shifting.
HL is a particular case where the planarization can be achieved by a small amount of thermal energy, paving the way for a tunneling mechanism on the π bond-shifting. Therefore, SCT calculations explain the experimentally observed splitting of the 13C NMR signals at −160 °C (ref. 28) by a thermally activated tunneling process.
For tBu3PL, the expected behaviour is more similar to HL than to PL. The tBu groups must rotate to match the shifting π bonds, a process that cannot be achieved purely by QMT and requires thermal energy (another example of thermally activated tunneling). Me3PL has similar energy requirements as tBu3PL, but in this case the light hydrogens in the Me groups can still rotate by QMT.
We hope these predictions and elucidations on QMT will be put to the test by experimentalists' hands.
Acknowledgements
The author wish to thank the fruitful insights provided by Weston T. Borden, the abundant technical support of David A. Hrovat and the help and support of Mariela Pavan.Notes and references
- D. W. Whitman and B. K. Carpenter, J. Am. Chem. Soc., 1980, 102, 4272–4274 CrossRef CAS.
- D. W. Whitman and B. K. Carpenter, J. Am. Chem. Soc., 1982, 104, 6473–6474 CrossRef CAS.
- B. K. Carpenter, J. Am. Chem. Soc., 1983, 105, 1700–1701 CrossRef CAS.
- M. J. Huang and M. Wolfsberg, J. Am. Chem. Soc., 1984, 106, 4039–4040 CrossRef CAS.
- M. J. S. Dewar, K. M. Merz and J. J. P. Stewart, J. Am. Chem. Soc., 1984, 106, 4040–4041 CrossRef CAS.
- P. Čársky, R. J. Bartlett, G. Fitzgerald, J. Noga and V. Špirko, J. Chem. Phys., 1988, 89, 3008–3015 CrossRef PubMed.
- R. Lefebvre and N. Moiseyev, J. Am. Chem. Soc., 1990, 112, 5052–5054 CrossRef CAS.
- Kinetics and spectroscopy of carbenes and biradicals, ed. M. Platz, Plenum Press, New York, 1990 Search PubMed.
- A. Datta, D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 2008, 130, 6684–6685 CrossRef CAS PubMed.
- P. S. Zuev, R. S. Sheridan, T. V. Albu, D. G. Truhlar, D. A. Hrovat and W. T. Borden, Science, 2003, 299, 867–870 CrossRef CAS PubMed.
- X. Zhang, D. A. Hrovat and W. T. Borden, Org. Lett., 2010, 12, 2798–2801 CrossRef CAS PubMed.
- R. A. Moss, R. R. Sauers, R. S. Sheridan, J. Tian and P. S. Zuev, J. Am. Chem. Soc., 2004, 126, 10196–10197 CrossRef CAS PubMed.
- S. Kozuch, X. Zhang, D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 2013, 135, 17274–17277 CrossRef CAS PubMed.
- S. Kozuch, Phys. Chem. Chem. Phys., 2014, 16, 7718–7727 RSC.
- A. Toyota and S. Koseki, J. Phys. Chem., 1996, 100, 2100–2106 CrossRef CAS.
- M. J. Bearpark, L. Blancafort and M. A. Robb, Mol. Phys., 2002, 100, 1735–1739 CrossRef CAS.
- R. Haag, D. Schröder, T. Zywietz, H. Jiao, H. Schwarz, P. von Ragué Schleyer and A. de Meijere, Angew. Chem., Int. Ed. Engl., 1996, 35, 1317–1319 CrossRef.
- T. Bally, S. Chai, M. Neuenschwander and Z. Zhu, J. Am. Chem. Soc., 1997, 119, 1869–1875 CrossRef CAS.
- P. De Mayo, R. Bloch and R. A. Marty, J. Am. Chem. Soc., 1971, 93, 3071–3072 CrossRef CAS.
- M. Suda and K. Hafner, Tetrahedron Lett., 1977, 18, 2449–2452 CrossRef.
- K. Hafner, R. Dönges, E. Goedecke and R. Kaiser, Angew. Chem., Int. Ed. Engl., 1973, 12, 337–339 CrossRef.
- H. J. Dauben and D. J. Bertelli, J. Am. Chem. Soc., 1961, 83, 4659–4660 CrossRef.
- E. Le Goff, J. Am. Chem. Soc., 1962, 84, 3975–3976 CrossRef CAS.
- K. Hafner and H. U. Süss, Angew. Chem., Int. Ed. Engl., 1973, 12, 575–577 CrossRef.
- Z. U. Levi and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 2796–2797 CrossRef CAS PubMed.
- T. Lendvai, T. Friedl, H. Butenschön, T. Clark and A. de Meijere, Angew. Chem., Int. Ed. Engl., 1986, 25, 719–720 CrossRef.
- O. T. Summerscales and F. G. N. Cloke, Coord. Chem. Rev., 2006, 250, 1122–1140 CrossRef CAS PubMed.
- E. Vogel, H. Königshofen, J. Wassen, K. Müllen and J. F. M. Oth, Angew. Chem., Int. Ed. Engl., 1974, 13, 732–734 CrossRef.
- R. G. Bryant, J. Chem. Educ., 1983, 60, 933 CrossRef CAS.
- IUPAC Gold Book, http://goldbook.iupac.org.
- D. I. Lyakh, V. F. Lotrich and R. J. Bartlett, Chem. Phys. Lett., 2011, 501, 166–171 CrossRef CAS PubMed.
- D. G. Truhlar and B. C. Garrett, Annu. Rev. Phys. Chem., 1984, 35, 159–189 CrossRef CAS.
- A. Fernandez-Ramos, B. A. Ellingson, B. C. Garrett and D. G. Truhlar, in Rev. Comput. Chem., ed. K. B. Lipkowitz and T. R. Cundari, John Wiley & Sons, Inc., 2007, vol. 3, pp. 125–232 Search PubMed.
- J. Zheng, S. Zhang, B. J. Lynch, J. C. Corchado, Y.-Y. Chuang, P. L. Fast, W.-P. Hu, Y.-P. Liu, G. C. Lynch, K. A. Nguyen, C. F. Jackels, A. Fernandez Ramos, B. A. Ellingson, V. S. Melissas, J. Villà, I. Rossi, E. L. Coitiño, J. Pu, T. V. Albu, R. Steckler, B. C. Garrett, A. D. Isaacson and D. G. Truhlar, POLYRATE 2010-A: Computer Program for the Calculation of Chemical Reaction Rates for Polyatomics Search PubMed.
- J. Zheng, S. Zhang, J. C. Corchado, Y.-Y. Chuang, B. A. Ellingson, E. L. Coitiño and D. G. Truhlar, GAUSSRATE 2009-A Search PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci; G. A. Petersson; H. Nakatsuji; M. Caricato; X. Li; H. P. Hratchian; A. F. Izmaylov; J. Bloino; G. Zheng; J. L. Sonnenberg; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; J. A. Montgomery JrJ. E. Peralta; F. Ogliaro; M. Bearpark; J. J. Heyd; E. Brothers; K. N. Kudin; V. N. Staroverov; R. Kobayashi; J. Normand; K. Raghavachari; A. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; N. Rega; J. M. Millam; M. Klene; J. E. Knox; J. B. Cross; V. Bakken; C. Adamo; J. Jaramillo; R. Gomperts; R. E. Stratmann; O. Yazyev; A. J. Austin; R. Cammi; C. Pomelli; J. W. Ochterski; R. L. Martin; K. Morokuma; V. G. Zakrzewski; G. A. Voth; P. Salvador; J. J. Dannenberg; S. Dapprich; A. D. Daniels; Ö. Farkas; J. B. Foresman; J. V. Ortiz; J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2010.1, a package of ab initio programs, molpro, 2012 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 242–253 CrossRef.
- I. García Cuesta, S. Coriani, P. Lazzeretti and A. M. J. Sánchez de Merás, ChemPhysChem, 2006, 7, 240–244 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 CrossRef.
- B. C. Garrett and D. G. Truhlar, J. Chem. Phys., 1983, 79, 4931 CrossRef CAS PubMed.
- A. Pross, L. Radom and N. V. Riggs, J. Am. Chem. Soc., 1980, 102, 2253–2259 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SCT calculations on CBD, conformational energies of tBu3PL, tables of rates of reactions, geometries of the critical points, benchmark of the DFT method. See DOI: 10.1039/c4ra02191f |
| ‡ Strictly speaking, HL and APL are not antiaromatic since they are not planar.30 However, they still follow other criteria and characteristics of antiaromatic systems, such as the alternating single and double bonds. Interestingly, breaking the planarity rule is what makes the tunneling improbable, and therefore a purely antiaromatic molecule (such as PL) has a higher tendency to automerize by QMT. |
| § As a reviewer points out, the NMR experimental studies involve 13C/12C isotopomers, which provide slightly different – and hardly distinguishable – automerization rates (this is exemplified on PL in the ESI†). |
| This journal is © The Royal Society of Chemistry 2014 |