
Luminous block copolymer–quantum dots hybrids formed by cooperative assembly in a selective solvent†
Abstract
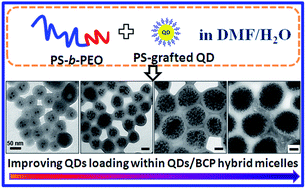
The formation of well-defined polymer/inorganic nanoparticle (NP) hybrid micelles is of considerable interest to the development of nanomaterials with desired optical, electric, magnetic, and mechanical properties. Herein, we introduce the efficient encapsulation of monodisperse polystyrene (PS)-grafted semiconductor quantum dots (QDs) into the PS cores of PS-b-PEO micelles through a “solution phase self-assembly” approach. We demonstrated that the size and the QD loading number of the coassemblies can be thermodynamically or kinetically tuned by varying the concentration of QDs and/or block copolymer (BCP) in the initial solution. Moreover, diverse morphologies of QD/BCP hybrid assemblies can be obtained by tuning the experimental parameters, such as the size of QDs, stirring time and many others. Interestingly, a novel Janus complex comprising a vesicular part without QDs and a spherical part with lots of QDs was firstly reported in our work. The Janus complexes were unstable and tended to transform their morphologies to the predominantly spherical QD-loaded micelles. The QD/BCP hybrid assemblies can present good water solubility, stability, lower toxicity and high luminous performance, indicating their potential applications in the biological field, especially for use as fluorescence probes and labels.
 Please wait while we load your content...
Please wait while we load your content...

