Deposition of Pd–Fe nanoparticles onto carbon spheres with controllable diameters and applied for CO catalytic oxidation
Weiliang Han,
Zhicheng Tang*,
Peng Zhang and
Gongxuan Lu
State Key Laboratory for Oxo Synthesis and Selective Oxidation, and National Engineering Research Center for Fine Petrochemical Intermediates, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, 730000, China. E-mail: tangzhicheng@licp.cas.cn; Fax: +86-931-8277088; Tel: +86-931-4968083
First published on 5th May 2014
Abstract
In this paper, a series of Pd–Fe/carbon sphere (CS) catalysts were prepared by a co-precipitation method and applied in low-temperature CO oxidation reactions. The effect of the particle sizes of carbon spheres, calcination temperatures of catalysts, Pd loadings and H2 reduction were investigated in detail. SEM and TEM characterizations of carbon spheres that were prepared by a hydrothermal method indicated non-porous structure, and the CSs surface was covered by Pd–Fe composites after the preparation of the catalyst. The XPS characterization of the catalysts showed that there were rich active oxygen species, more FeO(OH) species and more tetravalent Pd (PdO2) species on the surface of the Pd–Fe/CSs catalyst. These factors are expected be helpful for CO oxidation. When Pd loading was 1.0 wt%, the Pd–Fe/CSs (CSs were prepared by 1.0 mol L−1 glucose solution) catalyst, which was calcined at 200 °C without H2 reduction, had the highest activity.
1 Introduction
Catalytic oxidation of CO is of great importance because of its wide application such as air purification, CO gas sensors, vehicle exhaust pollution control,1–3 etc. Among all the catalysts for CO catalytic oxidation, Pd catalysts have attracted considerable attention because of their stability. However, the activity of Pd catalysts is lower than those of Au and Pt in CO oxidation.4–6 Thus, recent studies have focused on increasing the activity of Pd catalysts. Satsuma et al.5 also investigated the effect of supports (CeO2, TiO2, Al2O3, ZrO2, and SiO2) on CO oxidation over Pd catalysts, which indicated that the oxygen storage properties of metal oxide supports was one of the activity-controlling factors for CO oxidation. We prepared a series of Pd–Ce-supported ZSM-5 zeolite catalysts by a co-impregnation method, and found that the properties of ZSM-5 zeolite had a stronger influence on CO oxidation in the previous work.7 From these studies, it is clear that a support has an important influence on the catalytic performance for low-temperature CO catalytic oxidation.Carbon spheres (CSs) are widely used in adsorption, catalysis and catalyst supports because of their advantages of minimal surface energies and controllable sizes, morphologies, chemical properties and high mechanical stability.8–10 Various approaches have been used to prepare CSs such as chemical vapor deposition11,12 and high-temperature pyrolysis.13 However, these methods require sophisticated equipment, rigorous conditions or catalysts. Therefore, it is of great significance to explore energy-saving and cost-effective routes to fabricate CSs.
The hydrothermal method is unique because of its simple operational and mild reaction condition requirements. Since the first studies, interest in hydrothermal treatment for the preparation of CSs has increased.14–16 Wang et al.14 reported carbon microspheres with diameters of several micrometers synthesized by the hydrothermal treatment of methylcellulose sol at 400 °C, and discussed the formation mechanism of these spheres based on the reaction system features. Liu et al.15 prepared CSs with controllable sizes rich in oxygen-containing groups using a simple hydrothermal treatment of glucose. They also investigated the effects of the hydrothermal parameters, including the concentration of glucose, reaction temperature, duration, and the second hydrothermal treatment. Wang et al.16 successfully fabricated CSs with amino groups on their surface by the hydrothermal approach with ammonia in the precursor solution.
The Pd–Fe-based catalyst displays excellent activity on CO oxidation.17–19 Machida et al.17 reported that the bimetal nanoparticles of Pd–Fe were deposited onto CeO2 using a dual-arc plasma to study their structures and catalytic properties for CO oxidation. Their results showed that the bimetal nanoparticle catalysts exhibited a higher catalytic activity for CO oxidation, and the activity was further enhanced even after thermal aging at 900 °C in H2O/air. Lu et al.18 prepared Pd–Fe–Ox catalysts supported on SBA-15, CeO2 nanoparticles with rich (111) facets and CeO2 nanorods with rich (200) facets. Their results showed that when CeO2 nanorods were used as a support, Pd–Fe–Ox catalyst exhibited higher activity (T100 = 10 °C), resulting from the rich (200) facets of CeO2 nanorods, leading to the formation of a large number of oxygen vacancies on the surfaces of the Pd–Fe–Ox catalysts. Deng et al.19 found that the Pd catalysts supported on ferric-hydroxide were prepared by a co-precipitation method without calcination and exhibited high activity for CO oxidation.
These results suggest that Pd–Fe-based catalysts show superior activity for CO oxidation. However, Pd–Fe-based catalysts are powders, according to the aforementioned literatures, making it difficult to apply in practice. The loading of Pd–Fe onto a structure support is important for promoting the application of Pd–Fe-based catalysts. Carbon spheres have abundant surface oxygen species, including controllable sizes and regular shapes. The synthesizing process of carbon spheres is simple and inexpensive and may be a superior catalyst support. Thus, the deposition of Pd–Fe nanoparticles onto the carbon spheres is significant to the application of the Pd–Fe-based catalyst.
In this paper, CSs with controllable sizes were used as support, and a series of Pd–Fe/CSs catalysts were prepared successfully by the co-precipitation (CP) method and applied to low-temperature CO oxidation. The catalysts were characterized by XRD, XPS, BET, FE-SEM and HR-TEM in detail.
2 Experimental
2.1 Synthesis of CSs
A glucose solution (0.1–1.5 mol L−1, 80 mL) was transferred into a stainless-steel autoclave with a 60 mL capacity, and was autoclaved at 180 °C for 7 h. Then, the solution was cooled to room temperature. The black precipitate was collected and sequentially washed with water and anhydrous ethanol. The precipitate was then dried at 80 °C for 6 h. The brown solid obtained was identified as CSs.2.2 Preparation of catalyst
The Pd–Fe/CSs catalyst was prepared by the co-precipitation method. First, 0.5 g CSs were suspended in Na2CO3 solution. Pd(NO3)2 and Fe(NO3)3 solutions were then added to a separating funnel. When plug was opened, the mixed solutions of Pd(NO3)2 and Fe(NO3)3 flowed into a Na2CO3 solution containing the CSs support. The Na2CO3 solution was then added to adjust the pH and aged for 3 h. Finally, the solution was filtered and washed with distilled water. The resulting solid was dried at 60 °C overnight and subsequently calcined at 200 °C for 4 h. The Pd loading amount was adjusted from 0.3% to 1.0%, and the Fe loading amount was 50 wt%.The Pd–FeOx was prepared according to abovementioned method without CSs.
2.3 Catalyst characterization
Powder X-ray diffraction (XRD) analysis was performed to verify the crystallographic phases present in the carbon spheres. The XRD patterns of the samples were recorded on a Rigaku D/MAX-RB X-ray diffractometer with a target of Cu Kα operating at 60 kV and 55 mA with a scanning speed of 0.5° min−1 and a scanning-angle (2θ) range of 10–80°.The chemical states of the atoms in the catalyst surface were investigated by X-ray photoelectron spectroscopy (XPS) on a VG ESCALAB 210 Electron Spectrometer (Mg Kα radiation; hν = 1253.6 eV). The XPS data were calibrated using the binding energy of C 1s (284.6 eV) as the standard.
The surface morphologies of the carbon spheres were also determined by field emission scanning electron microscopy (FE-SEM) in combination with energy dispersive X-ray analysis on an ESEM-FEG/EDAX Philips JSM-6701F instrument operating at 20 kV using catalyst powders supported on carbon tape.
High-resolution transmission electron microscopy (HR-TEM) experiments were carried out to study the fine morphology of the Pd nanoparticles dispersed in the catalysts using a FEI TECNAIG2 Microscope operated at 200 kV.
The specific surface area and the mean pore diameter of the catalysts were determined by nitrogen adsorption in accordance with the BET method using a Micromeritics ASAP 2010 instrument. The BET surface area determinations were based on six measurements at the relative pressures of N2 in the range of 0.05–1.00.
2.4 Measurements of catalytic performance
Catalytic activity tests were performed in a continuous-flow, fixed-bed microreactor. A glass tube with an inner diameter of 6 mm was chosen as the reactor tube. Approximately 300 mg catalyst with an average diameter of 20–40 mesh was placed into the tube. The reaction gas mixture consisting of 1 vol% CO balanced with air was passed through the catalyst bed at a total flow rate of 50 mL min−1. A typical weight hourly space velocity (WHSV) was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1. The composition of the influent and the effluent gas was detected with an online GC-7890II gas chromatograph equipped with a thermal conductivity detector. As is well-known, a CO oxidation reaction is accompanied by a reduction in the number of moles. In this paper, this change in moles was neglected. Therefore, the CO conversion was calculated based on the outlet CO:
000 mL g−1 h−1. The composition of the influent and the effluent gas was detected with an online GC-7890II gas chromatograph equipped with a thermal conductivity detector. As is well-known, a CO oxidation reaction is accompanied by a reduction in the number of moles. In this paper, this change in moles was neglected. Therefore, the CO conversion was calculated based on the outlet CO: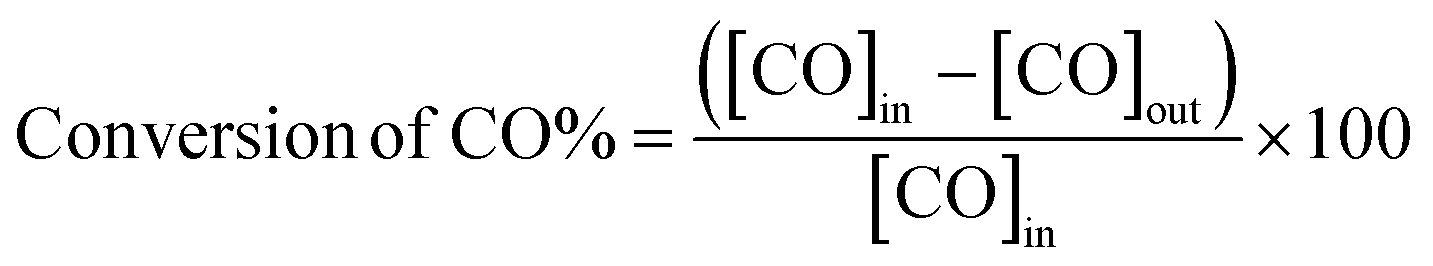
3 Results and discussion
3.1 Characterization of CSs
 | ||
| Fig. 1 SEM images and particle-size distribution of carbon spheres prepared by different glucose concentrations. (a) 0.1 mol L−1; (b) 0.5 mol L−1; (c) 1.0 mol L−1; (d) 1.5 mol L−1. | ||
 | ||
| Fig. 2 N2 adsorption–desorption isotherms of carbon spheres prepared by different glucose concentrations. (a) 0.1 mol L−1; (b) 0.5 mol L−1; (c) 1.0 mol L−1; (d) 1.5 mol L−1. | ||
| Sample | Glucose concentration (mol L−1) | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|---|
| a | 0.1 | 4.70 | 0.00781 | 6.65 |
| b | 0.5 | 3.11 | 0.00466 | 5.99 |
| c | 1.0 | 6.00 | 0.00769 | 5.13 |
| d | 1.5 | 4.65 | 0.00760 | 6.54 |
 | ||
| Fig. 3 TEM image of carbon sphere (a) and Pd–Fe/CSs catalyst (b), TEM-EDS mapping image taken from a square region (blue denotes Pd while yellow denotes Fe) of Pd–Fe/CSs catalyst (c), SEM image of Pd–Fe/CSs catalyst (d). | ||
3.2 The effect of carbon spheres size on the activity of CO oxidation
Fig. 4 shows the results of CO oxidation over the Pd–Fe/CSs catalysts with different particle sizes. The higher the reaction temperature was, the better were the catalytic activities, Fig. 4. The total conversion temperature (T100) was 75 °C, 85 °C, 45 °C, 50 °C and 65 °C corresponding to the catalysts of Pd–Fe/CSs (0.42 μm), Pd–Fe/CSs (0.63 μm), Pd–Fe/CSs (1.52 μm), Pd–Fe/CSs (4.95 μm) and Pd–FeOx, respectively. It was obvious that the addition of CSs could improve the activity for CO oxidation when the particle sizes of the CSs were 0.42 μm, 1.52 μm and 4.95 μm. As is well-known, a larger surface area of support not only is conducive to the loading and dispersal of Pd–Fe nanoparticles, but is also favorable for the adsorption and diffusion of reactants and products, all of which are helpful for CO oxidation. Comparing Fig. 2 and 4 and Table 1, it was easy to find that the activity improved along with an increase in the surface area of support. This implied that the activity was a direct factor in the surface area of support. However, samples a and d, which have almost identical surface areas, exhibited very different T100 values and evidenced that the surface area was not the only factor. The surface chemical species, surface active oxygen and distribution of active components may lead to significant differences in the catalytic activity. | ||
| Fig. 4 The effect of carbon spheres particle size on the activity of CO oxidation. (a) Pd–Fe/CSs (0.42 μm); (b) Pd–Fe/CSs (0.63 μm); (c) Pd–Fe/CSs (1.52 μm); (d) Pd–Fe/CSs (4.95 μm); and (e) Pd–FeOx. | ||
3.3 The effect of calcination temperature on the activity of CO oxidation
Fig. 5 shows the reaction results of CO oxidation over various Pd–Fe/CSs catalysts that were calcined at different temperature. Obviously, the Pd–Fe/CSs catalysts exhibited high activity for CO oxidation, and the reaction activity increased gradually with an increase in calcination temperatures at first and then decreased. It was observed that T100 were 45 °C, 40 °C, 35 °C and 70 °C for catalysts which were calcined at 60 °C, 150 °C, 200 °C and 300 °C, respectively. In order to further study the effect of calcination temperature on CO oxidation, all catalysts were characterized by XRD and XPS. | ||
| Fig. 5 The effect of calcination temperature on the activity of CO oxidation. | ||
Fig. 6 shows the XRD patterns of a series of catalysts that were prepared at different calcination temperatures. As is well-known, the palladium species was the active composition in the Pd catalyst. However, no characteristic diffraction peaks of the Pd particles appeared in all catalysts, which showed that the Pd species were highly dispersed. Diffraction peaks at 2θ = 18.5°, 30.8°, 35.2°, 43.0°, 57.5° and 62.7° were assigned to iron hydroxide (JCPDS PDF#22-0346), and it can be observed in Fig. 6a. In addition, with increasing calcination temperatures the intensity of iron hydroxide peaks decreased and the XRD peaks at 18.5° disappeared from Fig. 6b. It was clearly noticed in Fig. 6c that the peaks of iron hydroxide disappeared completely, one broad peak at 35.7° assigned to (211) crystalline plane of FeO(OH) was observed (JCPDS PDF#18-0639),21 which implied that Fe(OH)3 phases converted to FeO(OH). When the catalyst was heated to about 300 °C, two Fe2O3 species were observed in Fig. 6d. The peaks at 30.2°, 35.6°, 43.2° and 53.7° were assigned to maghemite-C (JCPDS PDF#39-1346), and other peaks at 24.1°, 33.1°, 35.6°, 40.8°, 49.4°, 54.1°, 57.6°, 62.4° and 64.0° were assigned to hematite (JCPDS PDF#33-0664). The calcination of the catalysts was a process of transformation from iron hydroxide to iron oxide. By comparing with the result of Fig. 5, it was observed that FeO(OH) species were helpful for the increase of activity.
 | ||
| Fig. 6 XRD patterns of Pd–Fe/CSs catalysts with different calcined temperatures. (a) 60 °C; (b) 150 °C; (c) 200 °C; (d) 300 °C. | ||
To demonstrate the active phases of the catalysts, which involved CO oxidation and variation of the surface species during the catalytic reaction in detail, XPS was used to investigate the chemical states of surface atoms in catalysts as well as the composition and related distribution of the surface elements. The Pd–Fe/CSs catalysts calcined at various temperatures were tested by XPS and the results are shown in Table 2 and Fig. 7.
| Sample | Calcined temperature (°C) | Oads/(Oads + Olatt) or (O′ + O′′)/(O + O′ + O′′) | Pd4+/(Pd4+ + Pd2+) | Fe content (%) | Pd content (%) |
|---|---|---|---|---|---|
| a | 60 | 0.472 | 0 | 9.83 | 0.11 |
| b | 150 | 0.516 | 0.218 | 9.98 | 0.12 |
| c | 200 | 0.530 | 0.423 | 10.81 | 0.13 |
| d | 300 | 0.336 | 0.762 | 13.28 | 0.12 |
 | ||
| Fig. 7 XPS spectra of Pd–Fe/CSs catalysts calcined at different temperatures. (a) 60 °C; (b) 150 °C; (c) 200 °C; (d) 300 °C. | ||
Fig. 7A shows the XPS spectra of Pd 3d for the Pd–Fe/CSs catalysts calcined at various temperatures. It was obvious that the peaks of Pd 3d shifted to the high binding energy with increases in calcination temperature. The Pd 3d spectrum shown in Fig. 7Aa was composed of a one spin–orbit split doublet, which was assigned to the bivalent Pd (PdO).22 With increases in calcination temperature, the tetravalent Pd (PdO2) appeared in Pd–Fe/CSs catalysts. The calculated percentages of tetravalent Pd (PdO2) are listed in Table 2. It is clear that the percentage of tetravalent Pd (PdO2) increased with increases in calcination temperature. By comparing with the result of Fig. 5, it was observed that PdO2 is the main species on the Pd–Fe/CSs catalysts.
Fig. 7B shows the XPS spectra of Fe 2p for the Pd–Fe/CSs catalysts calcined at different temperatures. When the catalysts were calcined, only one binding energy of Fe 2p appeared in the XPS spectra, but catalysts calcined at different temperatures had different binding energies. All the four catalysts had binding energies at 710.76 eV, 710.78 eV, 710.86 eV and 711.23 eV, corresponding to calcination temperatures of 60 °C, 150 °C, 200 °C and 300 °C, respectively.23 By comparing with the result of Fig. 6, Fe species of the catalysts calcined at 60 °C, 150 °C, 200 °C and 300 °C were Fe(OH)3, Fe(OH)3, FeO(OH) and Fe2O3, respectively. As is well-known, FeO(OH) could store more oxygen activity than Fe(OH)3 and Fe2O3.24 Thus, the catalyst calcined at 200 °C exhibited superior activity of CO oxidation.
Fig. 7C shows the XPS spectra of O 1s for the Pd–Fe/CSs catalysts calcined at different temperatures. In this case, three peaks (O, O′, O′′) could be identified by the deconvolution of the O 1s spectra. The binding energy at 529.85 eV (O) was assigned to O2− in the lattices of the catalyst, whereas the binding energy at 531.44 eV (O′) was assigned to surface adsorbed oxygen, such as O− or OH−.25,26 The binding energy above 533.29 eV (O′′) was associated with adsorbed molecular water.25,26 Usually, a higher oxygen species concentration on the catalyst surface is beneficial for the enhancement in catalytic activity.27 Thus, the relative content of the surface adsorbed oxygen species in the total surface oxygen can be estimated from the relative area of the sub peak, and the results are shown in Table 3. It was obvious that the relative content of the adsorbed oxygen species on the Pd–Fe/CSs catalyst calcined at 200 °C was the highest. As previously suggested, Pd–Fe/CSs catalyst calcined at 200 °C had a superior activity of CO oxidation.
| Sample | Catalyst | Oads/(Oads + Olatt) or (O′ + O′′)/(O + O′ + O′′) | Pd4+/(Pd4+ + Pd2+ + Pd0) | Fe content (%) | Pd content (%) |
|---|---|---|---|---|---|
| a | Pd–Fe/CSs | 0.803 | 0.597 | 16.98 | 0.330 |
| b | Pd–Fe/CSs-R | 0.489 | 0.263 | 8.26 | 0.200 |
3.4 The effect of Pd loading on the activity of CO oxidation
The effects of Pd loadings on the catalytic activity of the Pd–Fe/CSs catalysts for CO oxidation were investigated, and the results are shown in Fig. 8. When Pd loading was below 0.7%, CO conversion was clearly affected by Pd loading, which indicated that PdO2 nanoparticles were the active composition in the Pd–Fe/CSs catalysts. When Pd loading further increased, the CO conversion over the Pd–Fe/CSs catalyst exhibited a modest increase. In this case, the palladium nanoparticles agglomerated easily if the loading content of the precious metals was too high; this was not helpful for increasing catalytic activity. | ||
| Fig. 8 The effect of Pd-loading amounts on the activity of CO oxidation. | ||
3.5 The effect of H2 reduction on the activity of CO oxidation
In order to investigate the influence of pre-reductions on catalytic performance, the Pd–Fe/CSs catalysts (the optimal catalyst according to the abovementioned results) were further reduced at 50 °C in the stream of hydrogen gas with a rate of 30 mL min−1 for 1 h and were then cooled in hydrogen atmosphere to room temperature. It was shown that the performance of the reduced catalyst (Pd–Fe/CSs-R catalyst) was lower compared with the unreduced Pd–Fe/CSs catalyst (Fig. 9), which could be explained by the XPS results. | ||
| Fig. 9 Catalytic activity of the catalysts before (a) and after (b) H2 reduction for CO oxidation. | ||
The XPS spectra of Pd–Fe/CSs and Pd–Fe/CSs-R are presented in Fig. 10 and Table 3. Fig. 10A shows the XPS spectra of Pd 3d for the Pd–Fe/CSs and Pd–Fe/CSs-R catalysts. The Pd 3d spectrum shown in Fig. 10Aa was composed of two doublets, (u0, u2), (u1, u3) corresponding to the emission from the spin–orbit split 3d3/2 and 3d5/2 core levels. The two doublets (u0, u2), (u1, u3) were assigned to bivalent Pd (PdO) and tetravalent Pd (PdO2), respectively. After H2 reduction, three doublets, (v0, v3), (v1, v4), (v2, v5) were observed from Fig. 10Ab, which were assigned to zerovalent (Pd0), bivalent Pd (PdO) and tetravalent Pd (PdO2), respectively.21 According to the results of Fig. 5 and 7, PdO2 is the main species on the Pd–Fe/CSs catalysts. The calculated percentages of PdO2 are listed in Table 2. It can be seen that the portions of the PdO2 component and the surface loading of Pd in the Pd–Fe/CSs catalyst was relatively high.
 | ||
| Fig. 10 XPS spectra of the catalysts before (a) and after (b) H2 reduction. | ||
The spectra shown in Fig. 10B revealed the characteristic spin–orbit splitting of the Fe (2p) core levels [Fe (2p3/2) and Fe (2p1/2)]. Here, attention was only concentrated on the Fe (2p3/2) core-level electrons because it provided the spatial arrangement of the iron species. From Fig. 10Ba, only one peak appeared at around 710.81 eV, which was assigned to the characteristic peaks of FeO(OH).22 After H2 reduction, the XPS peaks site Fe species had not been altered. Because the XPS was a surface-sensitive technique, it offered an insight regarding the surface enrichment of the FeO(OH) site. This surface enrichment of FeO(OH) was advanced oxidation sites on the catalyst surface. The percentages of FeO(OH) are listed in Table 2. It can be seen that the FeO(OH) surface component in the Pd–Fe/CSs catalyst was relatively high.
Fig. 10C shows the O (1s) core level XPS spectra of the Pd–Fe/CSs and Pd–Fe/CSs-R catalysts. As suggested above, the peak O at 529.85 eV (BE), O′ at 531.44 eV (BE) O′′ at 533.29 eV (BE) were assigned to O2− in the lattice of the catalysts, surface adsorbed oxygen, such as O− or OH− and adsorbed molecular water, respectively.25,26 The relative content of the surface adsorbed oxygen species in the total surface oxygen can be estimated from the relative area of the sub peak, and the results are shown in Table 3. It can be noticed that the relative content of the adsorbed oxygen species on the Pd–Fe/CSs catalyst was higher than that on the Pd–Fe/CSs-R catalyst.
The results showed that Pd species were the key factors influencing the catalytic performances of the Pd–Fe/CSs catalysts, and surface area, surface adsorbed oxygen concentration and reducibility were direct factors affecting the catalytic performance of Pd–Fe/CSs. From Fig. 1–4 and Table 1, CSs prepared by 1.0 mol L−1 glucose solution were observed to be the highest BET surface area. When CSs were used as support, the catalyst had a superior activity of CO oxidation, which was because the high surface area of support was helpful for the loading of Pd–Fe species.
According to the results of activity and characterization, it could be determined that PdO2 was the main species on the Pd–Fe/CSs catalysts, and amorphous FeO(OH) was more helpful for CO oxidation than crystal Fe(OH)3 and Fe2O3. A higher oxygen adsorbed species concentration on the catalyst surface was beneficial for the enhancement of catalytic activity. There was a relatively high pressure of O2 in this study. According to the related literature,28 the FeO(OH) makes large amounts of weakly adsorbed oxygen available at the support surface. This oxygen is then able to compete with the lattice oxygen. At low CO pressures, the competition of lattice oxygen on the Pd surface may occur. Therefore, for the reaction mechanism of CO oxidation, possibly adhering to the Langmuir–Hinshelwood + redox (see Fig. 11): (a) CO was first adsorbed onto the tetravalent Pd; (b) the adsorbed CO on the Pd species were more easily oxidized to CO2 by catching the lattice oxygen (route 1 and 2) or adsorbed oxygen (route 3), and moreover, the catalysts produced an oxygen vacancy; (c) the oxygen vacancy would be replenished by O2 of the reaction gas to form the new active oxygen species and complete the redox cycle. FeO(OH) is main oxygen storage site in the catalysts, and thus route 1 is the main route.
 | ||
| Fig. 11 Illustration of reaction pathway for CO oxidation over Pd–Fe/CSs. | ||
4 Conclusions
The catalytic properties of the Pd–Fe catalysts supported on carbon spheres (CSs) were prepared and investigated for CO oxidation. The carbon spheres with controllable sizes were synthesized by the hydrothermal method. The Pd–Fe/CSs catalysts were characterized by XRD, XPS, BET, SEM and TEM in detail. The experimental results revealed that the particle sizes of carbon spheres, calcination temperature of catalysts, Pd loadings and H2 reduction had an effect on the activity of CO oxidation. By comparing the activity results of catalysts with the results of characterization, following conclusions were made: (a) the bigger the surface area of the CSs, the higher the activity of the catalysts; (b) PdO2 was the main active species on Pd–Fe/CSs catalysts; (c) amorphous FeO(OH) was more helpful for CO oxidation than crystal Fe(OH)3 and Fe2O3; (d) a higher oxygen adsorption concentration on the catalyst surface was beneficial for the enhancement of catalytic activity. At 1.0 wt% Pd loading, the Pd–Fe/CSs catalyst, which was calcined at 200 °C without H2 reduction, had the highest activity.Acknowledgements
The financial support of The National Basic Research Program of China (2013CB933201), West Light Foundation of The Chinese Academy of Sciences, Engineering Laboratory of Gansu Development and Reform Commission and Opening Project of State Key Laboratory for Oxo Synthesis and Selective Oxidation is gratefully acknowledged.References
- L. C. Wang, Q. Liu, X. S. Huang, Y. M. Liu, Y. Cao and K. N. Fan, Appl. Catal., B, 2009, 88, 204–212 CrossRef CAS PubMed.
- M. S. Chen, Y. Cai, Z. Yan, K. K. Gath, S. Axnanda and D. W. Goodman, Surf. Sci., 2007, 601, 5326–5331 CrossRef CAS PubMed.
- S. M. Mcclure and D. W. Goodman, Chem. Phys. Lett., 2009, 469, 1–13 CrossRef CAS PubMed.
- Y. Z. Li, Y. Yu, J. G. Wang, J. Song, Q. Li, M. D. Dong and C. J. Liu, Appl. Catal., B, 2012, 125, 189–196 CrossRef CAS PubMed.
- A. Satsuma, K. Osaki, M. Yanagihara, J. Ohyama and K. Shimizu, Appl. Catal., B, 2013, 132–133, 511–518 CrossRef CAS PubMed.
- A. Tomita, K. Shimizu, K. Kato and Y. Tai, Catal. Commun., 2012, 17, 194–199 CrossRef CAS PubMed.
- W. L. Han, P. Zhang, Z. C. Tang and G. X. Lu, Process Saf. Environ. Prot., 2013 DOI:10.1016/j.psep.2013.04.003.
- H. Deng, X. L. Li, Q. Peng, X. Wang, J. P. Chen and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS PubMed.
- A. A. Deshmukh, S. D. Mhlanga and N. J. Coville, Mater. Sci. Eng., R, 2010, 70, 1–28 CrossRef PubMed.
- H. K. Yu, G. R. Yi, J. H. Kang, Y. S. Cho, V. N. Manoharan, D. J. Pine and S. M. Yang, Chem. Mater., 2008, 20, 2704–2710 CrossRef CAS.
- Z. Mehraban, F. Farzaneh and V. Dadmehr, Mater. Lett., 2009, 63, 1653–1655 CrossRef CAS PubMed.
- C. M. Lei, W. L. Yuan, H. C. Huang, S. W. Ho and C. J. Su, Synth. Met., 2011, 161, 1590–1595 CrossRef CAS PubMed.
- Q. Lin, M. Zheng, T. Qin, R. Guo and P. Tian, J. Anal. Appl. Pyrolysis, 2010, 89, 112–116 CrossRef CAS PubMed.
- Q. Wang, F. Cao, Q. Chen and C. Chen, Mater. Lett., 2005, 59, 3738–3741 CrossRef CAS PubMed.
- M. Li, W. Li and S. Liu, Carbohydr. Res., 2011, 346, 999–1004 CrossRef CAS PubMed.
- X. Wang, J. Liu and W. Xu, Colloids Surf., A, 2012, 415, 288–294 CrossRef CAS PubMed.
- S. Hinokuma1, Y. Katsuhara, E. Ando, K. Ikeue and M. Machida, Catal. Today, 2013, 201, 92–97 CrossRef PubMed.
- Y. Shen, G. Lu, Y. Guo, Y. Wang, Y. Guo, L. Wang and X. Zhen, Catal. Commun., 2012, 18, 26–31 CrossRef CAS PubMed.
- L. Q. Liu, B. T. Qiao, Y. D. He, F. Zhou, B. Q. Yang and Y. Q. Deng, J. Catal., 2012, 294, 29–36 CrossRef CAS PubMed.
- X. M. Sun and Y. D. Li, Angew. Chem., Int. Ed., 2004, 43, 3827–3831 CrossRef CAS PubMed.
- W. Han, P. Zhang, X. Pan, Z. Tang and G. Lu, J. Environ. Chem. Eng., 2013, 1, 189–193 CrossRef CAS PubMed.
- K. S. Kim, A. F. Gossmann and N. Winograd, Anal. Chem., 1974, 46, 197–200 CrossRef CAS.
- C. D. wagner, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-Ray Photoelectron Spectrocopy, Physical Electronics Inc, USA, 1995 Search PubMed.
- W. Han, P. Zhang, X. Pan, Z. Tang and G. Lu, J. Hazard. Mater., 2013, 263, 299–306 CrossRef CAS PubMed.
- Z. M. Liu, J. M. Hao, L. X. Fu and T. L. Zhu, Appl. Catal. B, 2003, 44, 355–370 CrossRef CAS.
- Z. Qu, F. Yu, X. Zhang, Y. Wang and J. Gao, Chem. Eng. J., 2013, 229, 522–532 CrossRef CAS PubMed.
- R. J. H. Voorhoeve, J. P. Remeika and D. W. Johnson, Science, 1973, 180, 62–64 CAS.
- E. Bekyarova, P. Fornasiero, J. Kašpar and M. Graziani, Catal. Today, 1998, 45, 179–183 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |