Magnetically Recoverable Catalysts Based on Polyphenylenepyridyl Dendrons and Dendrimers†
E. Yu. Yuzik-Klimovaa,
N. V. Kuchkinaa,
S. A. Sorokinaa,
D. G. Morganb,
B. Borisb,
L. Zh. Nikoshvilic,
N. A. Lyubimovac,
V. G. Matveevac,
E. M. Sulmanc,
B. D. Steind,
W. E. Mahmoude,
A. A. Al-Ghamdie,
A. Kostopoulouf,
A. Lappasf,
Z. B. Shifrina*a and
L. M. Bronstein*be
aA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov St., Moscow, 119991, Russia
bIndiana University, Department of Chemistry, Bloomington, IN 47405, USA. E-mail: lybronst@indiana.edu
cTver State Technical University, Department of Biotechnology and Chemistry, 22 A. Nikitina St, Tver, 170026, Russia
dIndiana University, Department of Biology, Bloomington, IN 47405, USA
eKing Abdulaziz University, Faculty of Science, Department of Physics, Jeddah, Saudi Arabia
fFoundation for Research and Technology-Hellas, Institute of Electronic Structure and Laser, Heraklion, 71110, Crete, Greece
First published on 16th May 2014
Abstract
Here, a systematic study of magnetite nanoparticle (NP) formation in the presence of functional polyphenylenepyridyl dendrons and dendrimers of different generations and structures (such as focal groups, periphery and a combination of phenylene and pyridyl moieties) has been reported. For certain dendron/dendrimer concentrations and structures, well-dispersible, multi-core, flower-like crystals are formed which display ferrimagnetic-like behavior. It is noteworthy that the least complex second generation polyphenylenepyridyl dendrons with a carboxyl focal group already allow formation of flower-like crystals. Magnetically recoverable catalysts were obtained via Pd NP formation in the dendron/dendrimer shells of magnetite NP and tested in selective hydrogenation of dimethylethynylcarbinol to dimethylvinylcarbinol. Dependences of catalytic activity and selectivity on the dendron/dendrimer generation and structure, type of Pd species, and Pd NP size have been demonstrated. High selectivity and activity of these catalysts along with easy catalyst recovery and successful repeated use make them promising in catalytic hydrogenation.
Introduction
Magnetically recoverable catalysts have attracted considerable attention due to the combination of catalytic properties with easy catalyst recovery from reaction mixtures and their repeated use.1–5 Magnetically recoverable catalysts allow conservation of energy, more environmentally friendly processes and cheaper target products.6–12 Normally, these catalysts consist of functionalized magnetic nanoparticles (NP). Among various magnetic materials, magnetite (Fe3O4) is often preferred due to its strong magnetic response and stability in ambient conditions.13–20 Functionalization of these NP can be carried out in numerous ways involving surfactants, polymers, or other functional capping molecules.21–31 In the majority of cases, it is post-syntheses functionalization, i.e., iron oxide NP are first synthesized and then functionalized either by ligand exchange reaction32,33 or by placement of polymer chains on the nanoparticle surface,21,22 or by covalent attachment of functional molecules to the NP surface.34–36 In all functionalization protocols, however, it is difficult to control the distribution of functional ligands on the NP surface. From this point of view the most straightforward method allowing uniform distribution of functional capping molecules on the NP surface is direct formation of magnetic NP in the presence of functional ligands37 followed by incorporation of catalytic species in the ligand shells.In our preceding paper, a family of new polyphenylenepyridyl dendrons of different structures has been developed.38 As a proof of concept for the second generation dendrons with dicarboxylate focal points, it was proved that they can serve as capping molecules for magnetically recoverable catalysts. In the current paper, a systematic study of magnetite NP formation in the presence of dendrons and dendrimers of different generations and structures is presented. Magnetically recoverable catalysts were obtained via Pd NP formation in the dendron/dendrimer shells of iron oxide NP and tested in selective hydrogenation of dimethylethynylcarbinol (DMEC) to dimethylvinylcarbinol (DMVC). The dependences of catalytic activity and selectivity on the dendron/dendrimer generation and structure, type of Pd species, and the Pd NP size are demonstrated. High selectivity and activity of these catalysts along with easy catalyst recovery and successful repeated use make these nanomaterials promising for hydrogenation and likely for a number of other catalytic reactions.
Experimental part
Materials
Iron(III) acetylacetonate (99+%) was purchased from Acros Organics and was used as received. Palladium(II) acetate was purchased from Strem Chemicals and also used as received. DMEC (≥98%), toluene (99.8%), benzyl ether (≥98%), and 2-propanol (≥99.5%) were purchased from Sigma-Aldrich and used without purification. Acetone (99.5%) and chloroform (99.8%) were purchased from MACRON Chemicals and used as received.Synthetic procedures
The syntheses of functional polyphenylenepyridyl dendrons and dendrimers are described in our preceding papers.38,39 The dendrons and dendrimers used as capping molecules in this work are presented in Schemes 1 and 2. | ||
| Scheme 1 Structures of polyphenylenepyridyl dendrons of the second (1) and third (2 and 3) generations. Pyridine rings are shown in red, phenylene rings are in black, and carboxyl focal groups are shown in blue. | ||
 | ||
| Scheme 2 Structures of polyphenylenepyridyl dendrimers of the third generation with tetra- (4) and tri-substituted (5, 6) cores. Color coding is similar to that in Scheme 1. | ||
Iron oxide NP syntheses
Syntheses of iron oxide nanoparticles in the presence of dendrons/dendrimers were carried out according to the following protocol. In a typical experiment, the three-neck round-bottom flask (with elongated necks) equipped with a magnetic stir bar, a reflux condenser, and two septa, one of which contained an inserted temperature probe protected with a glass shield and the other had a long inserted needle, was loaded with 0.353 g (1 mmol) of Fe(acac)3, 0.140 g (0.11 mmol) of dendron 1, and 7 mL of benzyl ether. The flask was placed in a Glas-Col heating mantle attached to a digital temperature controller which in turn was placed on a magnetic stirrer. The flask was degassed by argon bubbling for 30 min under stirring. Then the temperature was raised to 60 °C at 10° min−1 and kept under stirring at this temperature for 30 min to allow solubilization. Then the temperature was increased with a heating rate 10° min−1 and the flask was allowed to reflux for 1 h at 283–285 °C. The flask was then removed from the heating mantle and allowed to cool to room temperature. To isolate NP, a part of the reaction solution was precipitated by ethanol and washed several times with ethanol and acetone until colorless and transparent supernatant was obtained. The latter was removed, while the precipitate was dispersed in chloroform upon sonication for 20 min. After that the solution was centrifuged for 7–10 min to remove aggregates if any. The rest of the reaction solution was stored in a refrigerator and was stable for many months. This sample is notated NP1-4. “NP” stands for iron oxide NP; the first number indicates a dendron/dendrimer according to Schemes 1 & 2 and the second number indicates the synthesis number according to Table 1. This table lists the reaction conditions and the sizes of the iron oxide NP formed.| Notation | Dendron/dendrimer loading (g) | Dendron/dendrimer loading (mmol) | Dendron/dendrimer concentration (mg mL−1) | Yield of NP (%) | Yield of aggregates (%) | Size of flower-like NP (nm) | Standard deviation of NP sizes (%) | Yield of flower-like NP (%) |
|---|---|---|---|---|---|---|---|---|
| a 0.353 g (1 mmol) Fe(acac)3 and 7 mL of benzyl ether were used in all procedures.b The iron oxide NP are fully aggregated. | ||||||||
| NP1-1 | 0.279 | 0.22 | 40 | 72 | 10 | 22.7 | 12.8 | 63.0 |
| NP1-2 | 0.837 | 0.66 | 120 | 5 | 83 | 77.3b | 18.0 | — |
| NP1-3 | 1.395 | 1.1 | 199 | 2 | 87 | 52.6b | 22.6 | — |
| NP1-4 | 0.140 | 0.11 | 20 | 75 | 8 | 21.8 | 15.1 | 61.1 |
| NP1-5 | 0.07 | 0.055 | 10 | 70 | 13 | 20.5 | 17.1 | 59.3 |
| NP2-1 | 0.616 | 0.22 | 88 | 0 | 93 | 55.8b | 16.1 | 100.0 |
| NP2-2 | 0.308 | 0.11 | 44 | 65 | 17 | 21.1 | 20.4 | 74.0 |
| NP2-3 | 0.154 | 0.055 | 22 | 68 | 15 | 16.1 | 20.5 | 42.7 |
| NP3-1 | 0.615 | 0.22 | 88 | 65 | 17 | 23.1 | 11.0 | |
| NP4-1 | 0.607 | 0.055 | 87 | 61 | 18 | 22.6 | 24.8 | 85.5 |
| NP5-1 | 0.592 | 0.073 | 85 | 59 | 16 | 34.4 | 19.5 | 76.0 |
| NP5-2 | 0.296 | 0.0365 | 42 | 63 | 17 | 32.0 | 20.6 | 89.6 |
| NP6-1 | 0.596 | 0.073 | 85 | 0 | 92 | 101.2b | 22.8 | — |
| NP6-2 | 0.397 | 0.049 | 57 | 0 | 90 | 105.4b | 20.7 | — |
| NP6-3 | 0.297 | 0.037 | 42 | 0 | 93 | 57.5b | 20.4 | — |
Interaction of iron oxide NP coated by dendrons/dendrimers with palladium acetate
The iron oxide NP solution (0.5 mg mL−1) was placed in a flask and purged with argon under stirring. The 1 mg mL−1 solution of Pd acetate in chloroform was also purged with argon and added to the iron oxide NP solution dropwise. This addition was notated I. The weight ratio of Pd acetate to iron oxide NP was 2/3. After addition the flask was closed and kept under stirring overnight at room temperature. The NP were collected with magnetic separation using a rare earth magnet and washed with chloroform 2–3 times. Then the sample was dried for 30 min in a vacuum oven and dispersed in 5 mL of ethanol under sonication. Alternatively, the Pd acetate solution was added to the NP solution at once, sonicated for 10 min and stirred overnight at room temperature. This procedure was notated II.Reduction of Pd complexes with hydrogen
Ethanol dispersion (5 mL) of Pd-containing sample was placed in a three-neck round-bottom flask equipped with a stir bar, a septum with a long needle, and a reflux condenser. The ethanol solution was diluted with 5 mL of distilled water. Then the reaction solution was purged with argon under stirring for 1 h and then bubbled with hydrogen under stirring for 2 h. After that the solution was transferred to a vial, the catalyst was separated with a magnet and washed several times with ethanol. Then the sample was dried in a vacuum oven overnight and dispersed in chloroform.Characterization
Electron-transparent NP specimens for transmission electron microscopy (TEM) were prepared by placing a drop of dilute solution onto a carbon-coated Cu grid. Images were acquired at an accelerating voltage of 80 kV on a JEOL JEM1010 transmission electron microscope. Images were analyzed with the National Institute of Health developed image-processing package ImageJ to estimate NP diameters. Between 150 and 300 NP were used for this analysis. High resolution TEM (HRTEM) images and energy dispersive X-ray spectra (EDS) were acquired at accelerating voltage 300 kV on a JEOL 3200FS transmission electron microscope equipped with an Oxford Instruments INCA EDS system. The same TEM grids were used for both analyses.Fourier Transform Infrared (FTIR) spectra were recorded on a Nicolet 510P FT-IR spectrometer. The samples were prepared by evaporating their chloroform solutions on a KBr disk.
X-ray diffraction (XRD) patterns were collected on an Empyrean from PANalytical. X-rays were generated from a copper target with a scattering wavelength of 1.54 Å. The step-size of the experiment was 0.02.
Thermal gravimetric analysis (TGA) was performed on TGAQ5000 IR manufactured by TA Instruments under nitrogen. About 2–3 mg of NP were placed into a 100 μL platinum pan by consecutive evaporations of small volumes of concentrated NP solutions. The experiments were carried out upon heating to 1000 °C with a rate of 10.0° min−1 with the purpose of determining the amount of dendron/dendrimer on the NP surface.
The magnetic properties of the samples were studied by a Superconducting Quantum Interference Device (SQUID) magnetometer (Quantum Design MPMS XL5). The measurements have been performed in dried nanoparticles on cotton in a gelatin capsule. The isothermal hysteresis loops, M(H), were measured at fields −1 ≤ H ≤ +1 Tesla. The dc magnetic susceptibility as a function of temperature, χ(T), was attained down to 5 K under zero-field cooled (ZFC) and field-cooled (FC) protocols, at H = 50 Oe.
Catalytic studies
Catalytic testing was carried out in a 60 mL isothermal glass batch reactor installed in a shaker and connected to a gasometric burette (for hydrogen consumption control). The total volume of liquid phase was 30 mL. Reaction conditions were: ambient hydrogen pressure, stirring rate 850 shakings/minute, temperature 90 °C. Toluene was used as a solvent.Samples were periodically taken and analyzed via GC-MS (Shimadzu GCMS-QP2010S) equipped with a capillary column HP-1MS (30 m × 0.25 mm i.d., 0.25 μm film thickness). Helium was used as a carrier gas at flow rate 1 mL min−1. Analysis conditions: oven temperature 60 °C (isothermal), injector and interface temperature 280 °C, ion source temperature 260 °C, range from 10 up to 200 m/z. For catalyst recycling, the catalyst was separated using a rare earth magnet, washed with toluene and ethanol and dried in vacuum oven for 3 hours.
Results and discussion
To assess the influence of dendron/dendrimer structures on the morphology of iron oxide NP, the dendrons of the second and third generations with the carboxyl focal group and different interior structure and the dendrimers of the third generation with different central cores and exterior (Schemes 1 and 2) were explored. Fig. 1 shows that iron oxide NP prepared in the presence of dendron 1 have a mixed morphology. The sample consists of multicore, flower-like NP and single core NP. The goal was to identify the proper conditions and the type of capping molecules to obtain iron oxide samples with a flower-like morphology only, as more interesting magnetic properties were expected.40 | ||
Fig. 1 TEM images of the Fe3O4 NP formed in the presence of 1 at the molar ratios Fe precursor/dendron 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.22 (a), 1:0.66 (c), 1:1.1 (d), 1:0.11 (e), and 1:0.055 (f). Insets in (a, e and f) show higher magnification images. HRTEM image and the corresponding FFT pattern (inset) of the NP of panel (a) are shown in (b). :0.22 (a), 1:0.66 (c), 1:1.1 (d), 1:0.11 (e), and 1:0.055 (f). Insets in (a, e and f) show higher magnification images. HRTEM image and the corresponding FFT pattern (inset) of the NP of panel (a) are shown in (b). | ||
The molar ratio of the iron precursor to capping molecules has been varied to assess its influence on the iron oxide NP formation. It is worth noting that the lower the dendron/dendrimer generation, the shorter the syntheses of these capping molecules, thus, the compounds are less expensive. In addition, the higher the Fe(acac)3/dendron(dendrimer) molar ratio, the lower the fraction of valuable dendrons/dendrimers used in syntheses, making this approach appealing for practical applications. Therefore, the possibility of obtaining a single morphology iron oxide NP with the second generation dendron 1 with a carboxyl focal group and the mixed pyridine–phenylene periphery (Scheme 1) has been explored.
Morphology and composition of iron oxide NP formed in the presence of dendron 1
At the molar ratio precursor/dendron 1:0.22 which was typically used in similar protocols,38 a mixture of multi-core and single core NP was obtained (Fig. 1a). The Fast Fourier Transform (FFT) pattern of the HRTEM image (inset in (b)) reveals the single crystalline arrangement inside the particles. Apparently, the multicore, flower-like NP were formed due to oriented attachment upon single core aggregation.41 The XRD pattern (Fig. S1, the ESI†) confirmed the magnetite phase which is consistent with our previous results.38 Thus, it was assumed that the dendron 1 functionality might be not sufficient to efficiently stabilize flower-like iron oxide NP at this concentration.
In order to increase the fraction of flower-like NP, the precursor/dendron molar ratio was increased to 1:0.66 and 1:1.1, i.e., ratios used when traditional surfactants (oleylamine and oleic acid) are employed.42 However, only aggregated NP were obtained and the size increases to 52.6 nm at molar ratio 1:1.1 (Fig. 1d) and to 77.3 nm at 1:0.66 (Fig. 1c). Alternatively, when the fraction of the dendrons decreases to molar ratios equal to 1:0.11 and 1:0.055 (Fig. 1e and f), the aggregation is minimal, while NP sizes are 21.8 nm and 20.5 nm, respectively, indicating that the dendron loading influences strongly the final morphology and size of the flower-like NP.
This behavior, when iron oxide NP aggregation occurs at the higher capping molecule loadings instead of better NP stabilization, can be attributed to the dendron aggregation at higher concentrations leading, in turn, to the formation of larger iron oxide NP and their subsequent aggregation.
The dendron 1 adsorption on the NP surface was further supported by the FTIR study. The FTIR spectra of dendron 1 and NP1-1 (see Fig. S2 and the text underneath, ESI†) show that the dendrons are present in the NP1-1 sample, although the majority of the dendron FTIR bands become weaker or nearly disappear as in the case of the bands responsible for vibrations of substituted arenes and pyridine rings. This might occur due to dendron adsorption on the iron oxide NP surface. A TGA trace of NP1-1 (Fig. S3, ESI†) shows a weight loss of 56% at 1000 °C due to dendron loss. It is noteworthy, however, that 1 itself decomposes with the formation of 32 wt% of coke (Fig. S3, ESI†) but the coke formation can be different in NP1-1due to iron oxide catalytic influence during thermal decomposition. Thus, the exact amount of dendrons on the NP surface cannot be determined, but it is estimated that it is not less than 56 wt%.
Magnetic measurements were carried out for NP1-1 and NP1-4, prepared with different amounts of dendron 1. The blocking temperature, TB, as indicated from the zero field and field cooled susceptibility curves (Fig. 2a) and the coercive field, HC, (Fig. 2b and c) are equal for the two samples due to the similar NP size. The TB for both samples is above 300 K, revealing that these NP are ferrimagnetic at room temperature. This is also in agreement with the non-zero coercive field, HC, derived from the isothermal magnetization curves at the same temperature presented in Fig. 2b. This holds promise for easy magnetic switching from ferrimagnetic to superparamagnetic behaviour at a minor temperature change and therefore controllable magnetic separation.
 | ||
| Fig. 2 ZFC-FC susceptibility curves as function of temperature under a magnetic field of 50 Oe (a) and isothermal magnetization curves at 300 K (b) and 5 K (c) for NP1-1 (red) and NP1-4 (black). In (a) ZFC curves are shown with filled symbols, while FC curves are shown with empty symbols. | ||
Thus, the least expensive, second generation polyphenylenepyridyl dendrons with a carboxyl focal group allow successful formation of flower-like crystals, but it was desirable to increase the multicore NP fraction.
Influence of the dendron/dendrimer structure and generation on the iron oxide NP morphology
In order to assess the influence of the dendron generation on iron oxide NP formation, the third generation dendron 2 whose structure is similar to that of 1 has been explored. At the molar ratio precursor/dendron 1:0.22 (dendron concentration = 88 mg mL−1) similar to those of NP1, large flower-like NP were formed with no single-core particles left out of the aggregate (Fig. S4. ESI†). Much smaller flower-like particles were obtained for decreased molar ratios (1:0.11 and 1:0.055) (Table 1). The fraction of single core iron oxide NP increases with the decrease of the dendron loading. On the other hand, for the third generation dendron 3, which has fully phenylene middle layer (with no pyridine moieties) and thus lower functionality, at the molar ratio of Fe to the dendron 1:0.22 (at 88 mg mL−1 concentration) mainly well-dispersible iron oxide NP (a mixture of multi-core, flower-like NP and single cores) were obtained. This indicates that both the generation and functionality of the dendrons play a crucial role in controlling the size and the morphology of the iron oxide NP formed.
To evaluate the importance of the dendron focal groups and the increase of the amount of functional groups in one molecule, iron oxide NP formation in the presence of third generation dendrimers (with structures 4, 5, 6 presented in Scheme 2) was studied.39 These macromolecules contain no carboxyl focal groups. Considering that a third generation dendron is a quarter of the dendrimer 4 with a tetra-substituted core, the molar ratio of the precursor to dendrimer was chosen 1:0.055 equal to the 1:0.22 ratio used for dendron capped iron oxide NP (Table 1). The iron oxide NP in NP4-1 (Fig. S5a, ESI†) were compared with those in NP2-1 (Table 1) because of similar interior and exterior structure (i.e., mixed phenylene and pyridyl periphery) in 2 and 4. The comparison of the iron oxide NP morphology shows that dendrimers allow much smaller flower-like NP without substantial aggregation. Thus, the higher total amount of functional groups and/or the presence of focal carboxyl groups facilitate interaction between dendrons and leads to the formation of larger particles and aggregates.
To evaluate the influence of the core structure, dendrimer 5 with similar periphery and a tri-coordinate core at the two different Fe/dendrimer molar ratios: 1:0.073 and 1:0.365 (Fig. S5, ESI†) was used. For 5, the ratio 1:0.073 is analogous to the 1:0.22 molar ratio of the dendron loading. TEM images presented in Fig. S5b and c (ESI)† demonstrate that for the dendrimers with the tri-coordinate core similar, although slightly larger particles were formed. Probably a larger size can be attributed to a more open the dendrimer 5 structure allowing weaker NP stabilization and therefore larger NP (Table 1). When the periphery of the dendrimer with a tri-coordinate core is fully pyridine (6), it strongly increases dendrimer intermolecular interactions leading to NP aggregation at all molar ratios used as shown in Table 1 (TEM images are not shown).
Formation of catalysts based on iron oxide NP stabilized by dendrons/dendrimers
To synthesize catalysts based on magnetic iron oxide NP, the latter were reacted with Pd acetate and further reduced using hydrogen as is shown in Scheme 3. The complexation of iron oxide NP with Pd species leads to the iron oxide NP aggregation due to interparticle complexation. This aggregation is crucial for easy magnetic separation.33,38 The reduction of PdAc-containing species in these aggregates with hydrogen resulting in Pd NP, does not change the aggregate size and morphology (Fig. 3).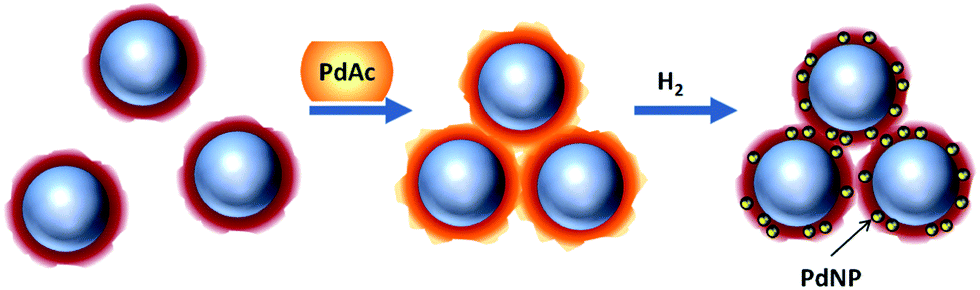 | ||
| Scheme 3 Schematic representation of the complexation of iron oxide NP (blue circles) coated with dendrons/dendrimers (dark red shell) with Pd acetate (PdAc, an orange shell) and the growth of Pd NP (yellow circles) in the iron oxide NP shells. | ||
 | ||
| Fig. 3 HRTEM image of NP1-1 after interaction with Pd acetate and Pd NP formation (NP1-1–PdNP). Yellow arrows indicate iron oxide NP, while red arrows point to the Pd NP. | ||
A dark-field STEM image and EDS mapping of the NP1-1–PdNP sample presented in Fig. S6 (ESI)† show superposition of Pd on Fe indicating that Pd NP are grown in the iron oxide NP dendron shells.
Pd NP are also incorporated in the shell of the iron oxide NP capped with dendrimer 4 (Fig. 4 and 5) as indicated from the elemental mapping (Fig. 5). In this case, Pd NP formed are even smaller (<1 nm). Thus, all dispersible flower-like iron oxide NP successfully interact with Pd acetate and allow formation of Pd NP, the sizes of which depend on the structure of the dendron/dendrimer.
 | ||
| Fig. 4 HRTEM image of NP4-1–PdNP. Smaller particles represent Pd NP (red arrows), while larger particles are the iron oxide NP (yellow arrows). | ||
 | ||
| Fig. 5 Dark-field STEM image (a) and Fe (b) and Pd (c) maps of the NP4-1–PdNP sample. Scale bar is 25 nm. | ||
The method of Pd acetate addition determines the final morphology of the aggregates. For the iron oxide NP capped with dendron 1, when the chloroform solution of Pd acetate was added slowly (dropwise) to the NP solution, the iron oxide NP self-assemble due to complexation into long rod-like aggregates (Fig. S7 and S8a, ESI†). When the iron oxide NP and Pd acetate solutions were mixed at once, the aggregates formed are irregular and fuzzier (Fig. S8b, ESI†). Alternatively, with the dendrons 3, rod-like Pd-containing aggregates are formed even upon fast Pd acetate addition, revealing that aligning of NP into rods is aided by a higher functionality of these dendrons (Fig. S9, ESI†).
For the particles based on 4, much smaller and fluffier aggregates are formed even upon slow addition of the Pd acetate solution to the NP solution (Fig. S11, ESI†). Such a different behavior upon complexation can be attributed to the presence of carboxyl groups in the exterior of the iron oxide NP stabilized with 3 and their absence for 4. Indeed, the FTIR spectrum of NP3-1 (Fig. S12, ESI†) shows a band at 1715 cm−1, indicating that not all carboxyl groups are adsorbed on the iron oxide NP surface which promotes self-assembly into rod-like aggregates.
Catalytic behavior of magnetically recoverable catalysts
Iron oxide NP containing Pd species were tested for their catalytic performance in a model reaction of selective hydrogenation of dimethylethynylcarbinol (DMEC) to dimethylvinylcarbinol (DMVC) (Scheme 4), which is an intermediate product in syntheses of fragrant substances and vitamins E and K.43 | ||
| Scheme 4 Hydrogenation of DMEC to DMVC. 2-Methylbutane-2-ol is a side product. “Cat” stands for the catalyst of hydrogenation. | ||
The data of catalytic testing are presented in Table 2. The turnover frequency (TOF) values in hydrogenation with the catalysts based on dendron 1 and prepared upon fast and slow addition of Pd acetate show that the latter leads to a slightly higher value, presumably, due to more accessible catalytic centers. At the same time, the catalyst based on 3 allows much higher activity (at the same selectivity) than the catalyst based on 1, indicating that aggregate morphology plays a minor role in overall catalytic properties. The major factors influencing the catalytic properties are likely the Pd NP size and the Pd NP surface functionalization due to the dendron/dendrimer adsorption. For example, in the case of NP3-1–PdNP, the Pd NP size is 1.5 nm, while for NP1-1–PdNP, the Pd NP diameter is 1.9 nm and the former catalyst is more active (Table 2).
| Dendron/dendrimer type | Pd state/content (%) | Notation of the catalyst | Pd NP size, nm | Standard deviation of NP sizes, % | TOF, s−1 | Selectivity at 95% of DMEC conversion, % |
|---|---|---|---|---|---|---|
| 1 | Pd NP/6.1 (PdAc added slowly) | NP1-1–PdNP-I | 1.8 | 25.1 | 2.2 | 97.5 |
| Pd NP/6.2 (PdA added fast) | NP1-1–PdNP-II | 1.9 | 26.2 | 2.5 | 97.9 | |
| 3 | PdAc/4.7 | NP3-1–PdAc | — | — | 4.3 | 97.5 |
| Pd NP/4.9 | NP3-1–PdNP | 1.5 | 18.3 | 9.3 | 97.5 | |
| 4 | PdAc/5.1 | NP4-1–PdAc | — | — | 1.6 | 97.0 |
| Pd NP/5.3 | NP4-1–PdNP | 0.9 | 23.4 | 6.7 | 96.8 | |
| 2nd use | 7.2 | 97.1 | ||||
| 3rd use | 7.1 | 97.0 | ||||
| Lindlar catalyst (2 wt% Pd in Pd/CaCO3) | — | — | — | 2.4 | 94.6 |
It is noteworthy, that the TOF is more than doubled when Pd NP are formed in the catalyst before catalytic reaction (NP3-1–PdNP) compared to the catalyst containing PdAc residues (NP3-1–PdAc). In the latter case, the Pd NP are also formed, but during a catalyst pretreatment step with hydrogen which is happening before catalytic reaction in a toluene solution (in a catalytic reactor). Toluene is a good solvent for dendrons/dendrimers and allows swelling of the iron oxide NP shells. As a result, Pd NP formed are much larger, 2.8 nm (Fig. S10, ESI†), compared to the 1.5 nm Pd NP formed by hydrogen reduction in ethanol–water mixture (NP3-1–PdNP). Thus, a clear dependence of TOF on the Pd NP size is observed. At the same time, the activity of NP4-1–PdAc was much lower than that of NP3-1–PdAc, indicating once more that aggregate morphology has nearly no influence on the catalytic performance. Alternatively, after hydrogen reduction of NP4-1–PdAc to NP4-1–PdNP TOF increases dramatically due to formation of 0.9 nm Pd NP (Fig. 6).
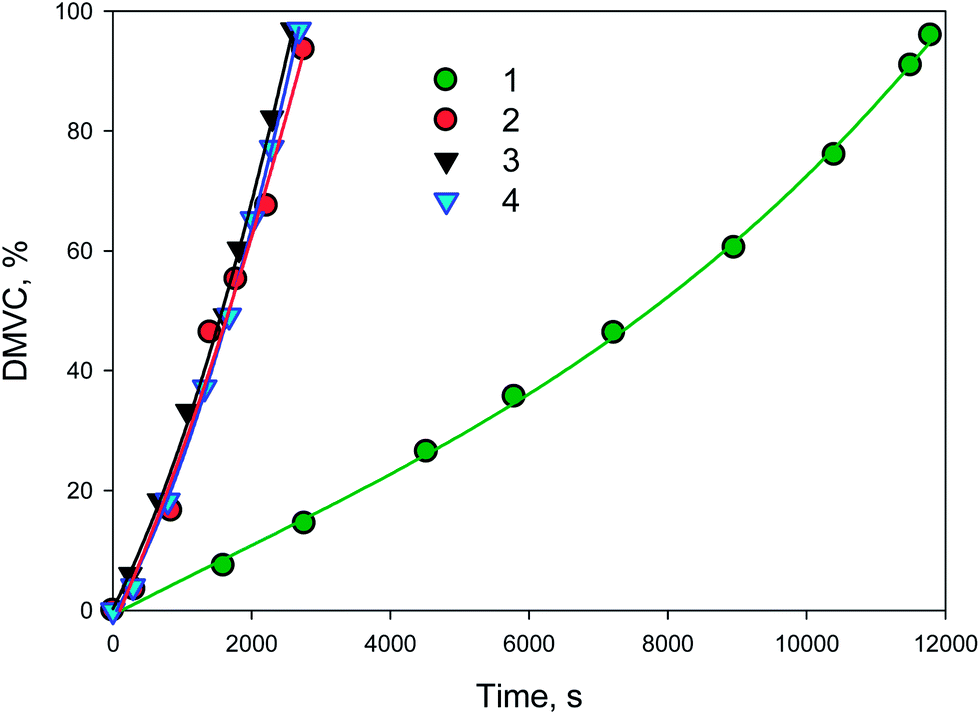 | ||
| Fig. 6 Kinetic curves of the DMVC accumulation with NP4-1–PdAc (1) and NP4-1–PdNP (2) as well as those for NP4-1–PdNP in the 2nd (3) and 3rd (4) uses. | ||
When NP4-1–PdNP was magnetically separated after hydrogenation and used again in two more catalytic cycles, the TOF and selectivity remained unaffected revealing exceptional stability of this catalyst (Table 2).
However, the lower activity of NP4-1–PdNP compared to that of NP3-1–PdNP was puzzling. The conventional wisdom is the smaller the NP, the higher the activity due to higher NP surface area. However, there are studies indicating that for the best catalytic performance, an optimal catalytic NP size is needed.44–47 Apparently, the Pd NP size of 0.9 nm in NP4-1–PdNP is below this optimal size, while the 1.5 nm Pd NP formed in NP3-1–PdNP should be close to the optimal size allowing the highest TOF demonstrated in this work. It is worth noting that the NP3-1–PdNP and NP4-1–PdNP catalysts are significantly more active and selective than the Lindlar catalyst (Table 2) which is traditionally used in a selective hydrogenation.38
Conclusions
Formation of flower-like multicore crystals in the presence of second and third generation polyphenylenepyridyl dendrons and third generation polyphenylenepyridyl dendrimers as capping molecules has been demonstrated. It was proven that the dendron/dendrimer structures and their concentrations control the iron oxide NP size, morphology and the tendency to aggregation. The higher the concentration of the capping molecules, the higher the probability of the aggregate formation and the larger the particle size. On the other hand, both the generation and functionality of the dendrons play a role in the size and morphology of iron oxide NP formed.With NP1-1 as an example, it was demonstrated that these NP are ferrimagnetic-like with a blocking temperature about 300 K, allowing easy magnetic switching. The interaction of these iron oxide NP with Pd acetate leads to the formation of magnetically recoverable aggregates. The hydrogen reduction of the Pd species results in Pd NP without affecting the aggregate size and morphology. It was also established that the catalytic properties in selective hydrogenation of DMEC are strongly influenced by the size of Pd NP, which, in turn, is determined by the structure of capping molecules. The highest TOF of 9.3 s−1 at the 97.5% selectivity is obtained for the catalyst based on dendron 3 and containing 1.5 nm Pd NP. Repeated uses (up to three catalytic cycles) of the magnetically recoverable catalyst demonstrate a stable catalytic performance making this class of catalysts promising for applications in various catalytic reactions.
Acknowledgements
The financial support of this work was provided in part by funding from the European Community's Seventh Framework Programme [FP7/2007–2013] under grant agreement no. CP-IP 246095, the NSF grant CHE-1048613, the Ministry of Education and Science of the Russian Federation and the Russian Foundation for Basic Research under grant numbers 12-03-31057, 14-03-31669 and 14-03-00876, and the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant no. (GR-33-7). L.B., W.M., and A.A. therefore acknowledge with thanks the DSR technical and financial support.Notes and references
- A.-H. Lu, E. L. Salabas and F. Schueth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed and references therein.
- B. Chang, Y. Tian, W. Shi, J. Liu, F. Xi and X. Dong, RSC Adv., 2013, 3, 20999–21006 RSC.
- Q. Dai, J. Wang, J. Yu, J. Chen and J. Chen, Appl. Catal., B, 2013, 144, 686–693 CrossRef PubMed.
- A. Bazgir, G. Hosseini and R. Ghahremanzadeh, ACS Comb. Sci., 2013, 15, 530–534 CrossRef CAS PubMed.
- D. Kundu, T. Chatterjee and B. C. Ranu, Adv. Synth. Catal., 2013, 355, 2285–2296 CrossRef CAS.
- Z. Wang, B. Shen, A. Zou and N. He, Chem. Eng. J., 2005, 113, 27–34 CrossRef CAS PubMed.
- Y. Zhu, S. C. Peng, A. Emi, Z. Su, Monalisa and R. A. Kemp, Adv. Synth. Catal., 2007, 349, 1917–1922 CrossRef CAS.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- L. M. Rossi, F. P. Silva, L. L. R. Vono, P. K. Kiyohara, E. L. Duarte, R. Itri, R. Landers and G. Machado, Green Chem., 2007, 9, 379–385 RSC.
- A. Saha, J. Leazer and R. S. Varma, Green Chem., 2012, 14, 67–71 RSC.
- B. R. Vaddula, A. Saha, J. Leazer and R. S. Varma, Green Chem., 2012, 14, 2133–2136 RSC.
- W. Sun, Q. Li, S. Gao and J. K. Shang, Appl. Catal., B, 2012, 125, 1–9 CrossRef CAS PubMed.
- D. H. Quinones, A. Rey, P. M. Alvarez, F. J. Beltran and P. K. Plucinski, Appl. Catal., B, 2014, 144, 96–106 CrossRef CAS PubMed.
- M. Shokouhimehr, J. E. Lee, S. I. Han and T. Hyeon, Chem. Commun., 2013, 49, 4779–4781 RSC.
- M. B. Gawande, V. D. B. Bonifacio, R. S. Varma, I. D. Nogueira, N. Bundaleski, C. A. A. Ghumman, O. M. N. D. Teodoro and P. S. Branco, Green Chem., 2013, 15, 1226–1231 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- M. S. A. Darwish, U. Kunz and U. Peuker, J. Appl. Polym. Sci., 2013, 129, 1806–1811 CrossRef CAS.
- Z. Yacob, A. Nan and J. Liebscher, Adv. Synth. Catal., 2012, 354, 3259–3264 CrossRef CAS.
- G. Marcelo, A. Munoz-Bonilla and M. Fernandez-Garcia, J. Phys. Chem. C, 2012, 116, 24717–24725 CAS.
- N. J. S. Costa, R. F. Jardim, S. H. Masunaga, D. Zanchet, R. Landers and L. M. Rossi, ACS Catal., 2012, 2, 925–929 CrossRef CAS.
- S. Wan, J. Huang, M. Guo, H. Zhang, Y. Cao, H. Yan and K. Liu, J. Biomed. Mater. Res., Part A, 2007, 80, 946–954 CrossRef PubMed.
- Y. Kang and T. A. Taton, Macromolecules, 2005, 38, 6115–6121 CrossRef CAS.
- B. D. Korth, P. Keng, I. Shim, S. E. Bowles, C. Tang, T. Kowalewski, K. W. Nebesny and J. Pyun, J. Am. Chem. Soc., 2006, 128, 6562–6563 CrossRef CAS PubMed.
- N. Nitin, L. E. W. LaConte, O. Zurkiya, X. Hu and G. Bao, JBIC, J. Biol. Inorg. Chem., 2004, 9, 706–712 CrossRef CAS PubMed.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759–1762 CrossRef CAS PubMed.
- Y. T. Lim, K. Y. Lee, K. Lee and B. H. Chung, Biochem. Biophys. Res. Commun., 2006, 344, 926–930 CrossRef CAS PubMed.
- Y.-W. Jun, Y.-M. Huh, J.-S. Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Yoon, K.-S. Kim, J.-S. Shin, J.-S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- H. A. Gussin, I. D. Tomlinson, D. M. Little, M. R. Warnement, H. Qian, S. J. Rosenthal and D. R. Pepperberg, J. Am. Chem. Soc., 2006, 128, 15701–15713 CrossRef CAS PubMed.
- Q. Zhang, S. Gupta, T. Emrick and T. P. Russell, J. Am. Chem. Soc., 2006, 128, 3898–3899 CrossRef CAS PubMed.
- G. Han, P. Ghosh and V. M. Rotello, Nanomedicine, 2007, 2, 113–123 CrossRef CAS PubMed.
- M. J. Hostetler, A. C. Templeton and R. W. Murray, Langmuir, 1999, 15, 3782–3789 CrossRef CAS.
- T. Gillich, C. Acikgoez, L. Isa, A. D. Schluter, N. D. Spencer and M. Textor, ACS Nano, 2013, 7, 316–329 CrossRef CAS PubMed.
- S. H. Gage, B. D. Stein, L. Z. Nikoshvili, V. G. Matveeva, M. G. Sulman, E. M. Sulman, D. G. Morgan, E. Y. Yuzik-Klimova, W. E. Mahmoud and L. M. Bronstein, Langmuir, 2013, 29, 466–473 CrossRef CAS PubMed.
- J. Babu, J. George and R. L. Varma, New J. Chem., 2013, 37, 2426–2432 RSC.
- K. Schadt, B. Kerscher, R. Thomann and R. Muelhaupt, Macromolecules, 2013, 46, 4799–4804 CrossRef CAS.
- M. Zeltner, R. N. Grass, A. Schaetz, S. B. Bubenhofer, N. A. Luechinger and W. J. Stark, J. Mater. Chem., 2012, 22, 12064–12071 RSC.
- K. Kumar, A. M. Nightingale, S. H. Krishnadasan, N. Kamaly, M. Wylenzinska-Arridge, K. Zeissler, W. R. Branford, E. Ware, A. J. de Mello and J. C. de Mello, J. Mater. Chem., 2012, 22, 4704–4708 RSC.
- N. V. Kuchkina, E. Y. Yuzik-Klimova, S. A. Sorokina, A. S. Peregudov, D. Antonov, L. Z. Nikoshvili, E. M. Sulman, D. G. Morgan, S. H. Gage, W. E. Mahmoud, A. A. Al-Ghamdi, L. M. Bronstein and Z. B. Shifrina, Macromolecules, 2013, 46, 5890–5898 CrossRef CAS.
- Z. B. Shifrina, M. S. Rajadurai, N. V. Firsova, L. M. Bronstein, X. Huang, A. L. Rusanov and K. Muellen, Macromolecules, 2005, 38, 9920–9932 CrossRef CAS.
- A. Kostopoulou, K. Brintakis, M. Vasilakaki, K. N. Trohidou, A. P. Douvalis, A. Lascialfari, L. Manna and A. Lappas, Nanoscale, 2014, 6, 3764–3776 RSC.
- M. Niederberger and H. Coelfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC.
- H. Zeng, P. M. Rice, S. X. Wang and S. Sun, J. Am. Chem. Soc., 2004, 126, 11458–11459 CrossRef CAS PubMed.
- W. Bonrath and M. Eggersdorfer, Catal. Today, 2007, 121, 45–57 CrossRef CAS PubMed.
- G.-F. Wei and Z.-P. Liu, Phys. Chem. Chem. Phys., 2013, 15, 18555–18561 RSC.
- S. Mukherjee, B. Ramalingam, L. Griggs, S. Hamm, G. A. Baker, P. Fraundorf, S. Sengupta and S. Gangopadhyay, Nanotechnology, 2012, 23, 485405–485418 CrossRef CAS PubMed.
- V. Viswanathan and F. Y.-F. Wang, Nanoscale, 2012, 4, 5110–5117 RSC.
- T. Yu, W. Wang, J. Chen, Y. Zeng, Y. Li, G. Yang and Y. Li, J. Phys. Chem. C, 2012, 116, 10516–10521 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD, TEM images, STEM and EDS maps, FTIR and TGA data. See DOI: 10.1039/c4ra00878b |
| This journal is © The Royal Society of Chemistry 2014 |