
The preparation of a novel polymeric sulfonium salt photoacid generator and its application for advanced photoresists†
Abstract
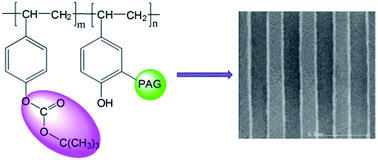
With poly(4-hydroxylstyrene) (PHS) as a raw material, a new type of polymeric photoacid generator (PAG) is synthesized with sulfonium perfluoroalkyl sulfonate groups bonded onto part of the benzene rings. The phenolic hydroxyl groups are esterified to increase the solubilities of the polymeric sulfonium salts in organic solvents. Upon irradiation to 254 nm light, the sulfonium units in the polymer effectively decompose to generate sulfonic acid. The photolysis is confirmed by UV spectroscopy and the generation of sulfonic acids which catalyze the decomposition of the t-BOC protection group of the polymeric PAGs. Making use of the polymeric PAGs, positive-tone chemically amplified 248-nm photoresists can be formed by them and ester acetal polymer with high acidolytic activity. The photolithography performance of the photoresist was evaluated using a KrF laser exposure system with high photosensitivity and resolution. This novel kind of polymeric sulfonium salt PAG is applicable for advanced chemically amplified resist materials.
 Please wait while we load your content...
Please wait while we load your content...

