
Fabrication of multi-functional PVDF/RGO composites via a simple thermal reduction process and their enhanced electromagnetic wave absorption and dielectric properties†
Abstract
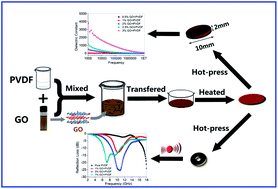
Polymer-composites of polyvinylidene fluoride (PVDF) and reduced graphene oxide (RGO) have been prepared from PVDF/GO membrane by a simple hot-molding technique. The specific interaction between oxygen-containing functional groups in the GO surface and fluorine groups in PVDF allows the GO to disperse into PVDF homogeneously and it can be thermally reduced into RGO after a hot-press process. Several characterizations such as AFM, XRD, FT-IR and Raman have been employed to confirm the thermally reduced process from GO to RGO in the composite. The enhanced absorption and dielectric properties were investigated; the results indicated that for the composites with a low filler loading of 3 wt%, the maximum reflection loss of the PVDF/RGO composite can reach −25.6 dB at 10.8 GHz, and the frequency bandwidth less than −10 dB is from 8.48 to 12.80 GHz, while, when the filler is 3 wt%, the dielectric constant can reach 3801 (103 Hz). The enhanced mechanism has been also explained in detail.
 Please wait while we load your content...
Please wait while we load your content...

