An insight into the extraction of transition metal ions by picolinamides associated with intramolecular hydrogen bonding and rotational isomerization†
Yan Lia,
Yiming Jiaa,
Zhenwen Wanga,
Xianghui Lia,
Wen Fenga,
Pengchi Deng*b and
Lihua Yuan*a
aCollege of Chemistry, Key Laboratory for Radiation Physics and Technology of Ministry of Education, Institute of Nuclear Science and Technology, Sichuan University, Chengdu 610064, China. E-mail: lhyuan@scu.edu.cn; Fax: +86-28-85418755; Tel: +86-28-85416996
bAnalytical & Testing Center, Sichuan University, Chengdu 610064, China. E-mail: pcdeng@yahoo.com
First published on 19th June 2014
Abstract
The clear connection between molecular structures of N-substituted picolinamides and extraction behaviour has been rationalized by highlighting the relationship between intramolecular hydrogen bonding and rotational isomerism. To this aim aromatic pyridine-2,6-dicarboxamides 1a–1c with N-substitution and their analogues 3a and 3b containing intramolecular hydrogen bonds were designed and synthesized. The results from the liquid–liquid extraction towards some representative transition metal picrates including Ag+, Hg2+, Pb2+, Cd2+, Zn2+, Cu2+, Co2+ and Ni2+ salts demonstrated that the higher selectivity and efficiency towards Hg2+ (88.6–95.4%) over other metal cations stem mainly from N-substitution via disruption of intramolecular H-bonding. X-ray structural analysis, and ordinary and variable-temperature proton and carbon NMR experiments provided supporting information for expounding the difference in extraction ability among these ligands, particularly the importance of N-substitution that leads to the formation of rotamers affecting the extraction process.
Introduction
Amide-based compounds and their corresponding metal complexes have been widely investigated due to the easy synthesis of ligands, high resistance to hydrolysis, and potential coordination ability of amide hydrogen and/or oxygens.1 These features render them find applications as extractants in separation technology,2 sensors in detecting metal ions,3 and building blocks for constructing architectures in catalysis.4 Diglycolamides,5 pyridine-modified calixarenes,6 CMPO-tripodands,7 and pyridine dicarboxyamides8 are examples of extraction systems that have demonstrated higher separation efficiency in discriminating lanthanides/actinides (Ln/Ac) elements. Among them, pyridine-based carboxyamides or dicarboxyamides and their analogues represent a class of ligands that have attracted attention in forming metal complexes9 and in use for metal separation.8,10 The amide linkages, as part of the molecular constituents of these ligands or complexes, were found to involve in interacting or coordinating with metal ions in synergy with the nitrogen of pyridine moiety.9,11 It has been noted that substitution on nitrogen of amide bonds of synthetic ligands led to improved performance in two-phase extraction of lanthanides/actinides elements.12 In fact, the effect of N-substituted ligands upon the extraction and separation efficiency had been observed in diamide systems ca. two decades before.13 However, the reason behind it is still not clearly clarified. For 2,6-dicarboxypyridine diamides, the explanation was limited to the electronic and steric effects that may dominate the atoms coordinating to the metal center in.14It was known that hydrogen bonding formation involving amide groups contribute a great deal to the construction of complex natural and artificial supramolecular assemblies.15 Incorporation of hydrogen bond (H-bond) groups into ligands was able to orient incoming groups or stabilize metal–ligand adducts.16 Besides the importance of metal complexing sites associated with amide linkage for effecting the separation process, intra- or intermolecular hydrogen bonding contained in the molecular structure of a ligand may also play a role in governing the extraction efficiency and selectivity. However, this aspect has scarcely been explored to date. We recently employed hydrogen bonded aromatic oligoamides with backbones preorganized by aid of intramolecular three-center hydrogen bonds for solvent extraction separation of transition metal ions.17 The importance of intramolecular hydrogen bonding present in these compounds was also demonstrated by the formation of their corresponding cyclo[6]aramides18 and efficiency in extraction towards Ln/An elements.19 Based on similar preorganization-induced folding mechanism, their higher aromatic amide polymers also exhibited selective extraction of thorium(IV) and rare earth elements.20 Very recently, we revealed that the subtle change of the coordination environment made by local intramolecular H-bonding of CMPO-modified calixarenes led to selective separation of light/heavy lanthanides and group separation between lanthanides and thorium/uranium.21 Despite much progress made in using 2,6-pyridine dicarboxyamides as solvent extractants22 and the well-known fact for the formation of amide rotamers,23 surprisingly, the correlation between the presence of intramolecular hydrogen bonding associated with amide NH in effecting liquid–liquid extraction behaviour and rotational isomerization is still unexplored.
With our continued interest in amide-based compounds and macrocyclic compounds for metal ion separation,24 we report herein on the exploration of intramolecular hydrogen bonding in regulating extraction process pertinent to rotational isomerism by N-substitution using a series of synthesized picolinamides. Transition metal ions including Ag+, Hg2+, Pb2+, Cd2+, Zn2+, Cu2+, Co2+, Ni2+ were selected to assess the outcome due to structural alternation of ligands used in the liquid–liquid extraction experiments.
Results and discussion
Initial consideration and molecular design
Picolinamides 1–3 were used in the present study (Scheme 1). Initially compound 1a bearing alkoxy substituents was designed as a control for ligand 2 in comparing extraction of transition metal ions. Compound 2 was reported to be potential extractant for minor actinides25 and palladium separation.26 Interestingly, examination of 1H NMR spectrum of 1a showed a complicated pattern comprising several sets of signals that cannot be designated as a pure component. However, a base peak at m/z 490.2701 corresponding to the most abundant species [M + H]+ in HRMS spectrum and a single peak from HPLC experiments excluded the possibility of the presence of any other impurities (see ESI†). Thus, the most likely possibility is the presence of rotational isomers for 1a since rotation around the amide bonds are considerably hindered upon introducing an ethyl group onto the amidic nitrogen. In other words, it is the coexistence of several conformational isomers that caused the complexity of its NMR spectrum. This led to the design of compound 3a without substitution on nitrogen atoms. At the same time, propoxy groups in 3a are placed at ortho-position adjacent to the amide bond to allow formation of intramolecular hydrogen bonds, thus partially rigidifying the backbone of the molecule (vide post). Free rotation around nitrogen of the amide NH and carbon of the phenyl ring is expected to be impossible. Compound 3b, which bears the same backbone with ortho-substituted methyl group, is designed to see if partial hydrogen bonding is still strong enough to maintain the molecular conformation as 3a. Positional isomers 1b and 1c are also designed for comparison. Given the importance of coordination directionality in forming extractive species, it can be envisioned that if the orientation of carbonyl oxygen atoms is manipulated by the presence of hydrogen bonding to restrict the amide rotations, different extraction behaviour should result. | ||
| Scheme 1 Synthesis of 2,6-pyridine dicarboxyamides 1a–1c, 2,25a 3a and 3b. | ||
Synthesis and solid state structures
Typically all 2,6-pyridine dicarboxyamides 1a–1c, 2, 3a and 3b were synthesized based on the coupling reactions of acyl chlorides and corresponding anilines according to Scheme 1. All of these compounds were characterized by 1H NMR, 13C NMR and HRMS. Compound 2 was prepared according to the reported procedure.25aThe key precursors 5a–5c were obtained by reaction of commercially available hydroxyl group-substituted nitrobenzene and propyl bromide in the presence of K2CO3. The syntheses of 6a–6c were carried out employing MeCN as ethylation agent.27 Treating the N-alkylated aniline derivatives 6a–6c with 4 resulted in 1a, 1b, and 1c in overall isolated yields of 82%, 92% and 88%, respectively. Compound 3a was readily prepared in 81% yield via two steps from hydrogenation of 5a with Pd/C as catalyst to afford 7, followed by coupling of 2,6-pyridinedicarbonyl dichloride 4.
Single crystals of ligands 1a and 3a were obtained by slow evaporation of a solution of CH2Cl2/n-hexane and ethyl acetate/n-hexane at room temperature, respectively. Selected bond lengths and angles for the two ligands from X-ray diffraction experiment are given in Table 1.
| Bond lengths (Ǻ) | Bond angles (°) | |||
|---|---|---|---|---|
| 1a | C3–C4 | 1.510(5) | C3–C4–O1 | 119.82(5) |
| O1–C4 | 1.222(7) | C3–C4–N2 | 117.63(2) | |
| N2–C4 | 1.349(0) | O1–C4–N2 | 122.53(2) | |
| N2–C5 | 1.474(5) | C4–N2–C5 | 119.12(8) | |
| N2–C7 | 1.431(5) | C4–N2–C7 | 122.92(6) | |
| C5–N2–C7 | 117.57(7) | |||
| 3a | C10–C11 | 1.507(8) | C11–C10–O2 | 121.75(7) |
| C10–O2 | 1.218(6) | C11–C10–N1 | 113.35(3) | |
| C10–N1 | 1.347(6) | O2–C10–N1 | 124.89(0) | |
| N1–H1 | 0.859(7) | C10–N1–H1 | 116.28(5) | |
| N1–C9 | 1.408(2) | C10–N1–C9 | 127.52(0) | |
| C16–C15 | 1.499(8) | H1–N1–C9 | 116.19(5) | |
| C16–O3 | 1.211(2) | C15–C16–O3 | 122.05(9) | |
| C16–N3 | 1.351(2) | C15–C16–N3 | 113.68(6) | |
| N3–H3 | 0.859(8) | O3–C16–N3 | 124.25(2) | |
| N3–C17 | 1.404(5) | C16–N3–H3 | 115.96(5) | |
| C16–N3–C17 | 128.08(5) | |||
| H3–N3–C17 | 115.95(0) | |||
Fig. 1a shows a view of the molecular structure of 1a. The molecule of 1a has a C2 symmetry, and the C2 axis passes through atoms C1 and N1 of the pyridine ring. The atoms of the amide groups and those connected to amide carbon and nitrogen (O1, C3, C4, N2, C5 and C7) are almost in a plane. The mean deviation from plane is 0.0401 Å. In addition, the bond angles across the amide nitrogen, C4–N2–C5, C4–N2–C7 and C5–N2–C7 are all approximately 120°. These data suggest the sp2 hybridization of the amide nitrogens N2, and the considerable double bond character for the OC–N amide bonds.11a,28 So far as the arrangement of amide nitrogen relative to pyridine nitrogen is concerned, the crystal structure of 1a reveals the anti–anti conformation: both of the amide nitrogens placed in trans position with respect to pyridine nitrogen. The dihedral angle between the pyridine ring and each of the amide planes is 64.99(5)°. To avoid n–n repulsion between the lone pairs of carbonyl oxygens, the two amide planes are staggered away from each other with a dihedral angle of 75.65(4)°. The orientation of the phenyl group is designated as E (trans) relative to the carbonyl oxygens when the OC–N amide bond is considered as a double bond. Consequently, 1a adopts E–anti–anti–E conformation (also see Scheme 2a, blue). The dihedral angle between the pyridine ring and each of the two phenyl groups (C7–C8–C9–C10–C11–C12) is 65.04(7)°.
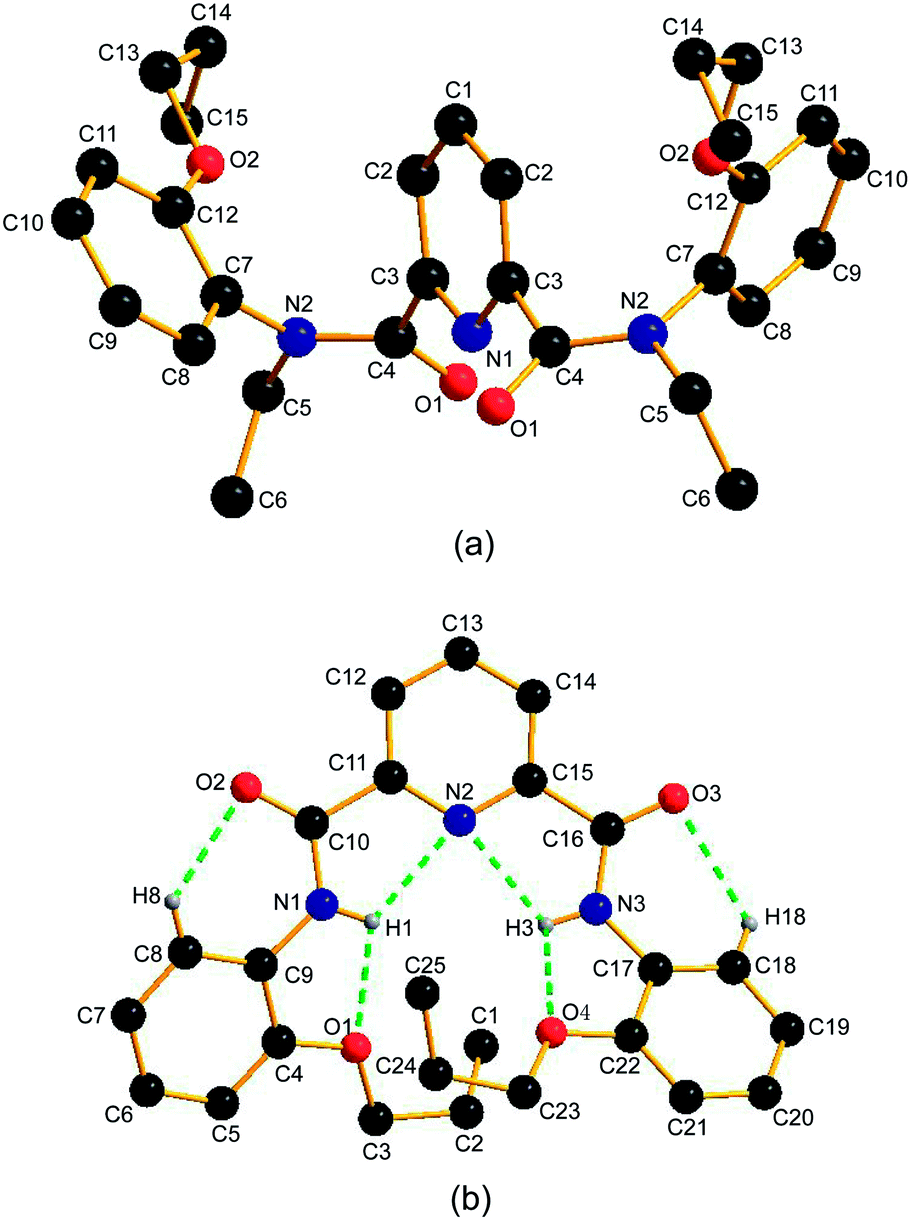 | ||
| Fig. 1 The crystal structures of (a) 1a and (b) 3a. Hydrogen atoms are omitted for the sake of clarity except for those forming hydrogen bonds. | ||
 | ||
| Scheme 2 Conformational conversion of isomers (a)–(c): (a) rotamer interconversion from 1a; (b) conformer formation of 3a via intramolecular H-bonding; (c) conformer formation of 3b via intramolecular H-bonding. (d) Conformation of compounds 2 (ref. 11a) and 9 (ref. 35) in solid state. | ||
For 3a, the solid state structure clearly indicates the presence of two intramolecular three-center hydrogen bonds, N2⋯H1⋯O1 and N2⋯H3⋯O4, each comprising two five-membered rings to fix the molecule in a crescent fashion as shown in Fig. 1b. The parameters of H-bonds involving in 3a are shown in Table 2. The H-bond lengths of N2⋯H1, O1⋯H1, N2⋯H3 and O4⋯H3 are 2.219(7) Å, 2.277(6) Å, 2.223(5) Å and 2.254(9) Å, respectively, suggesting the formation of strong H-bonds. Two additional weak hydrogen bonds O2⋯H8 and O3⋯H18 are also observed,29 the lengths of which are 2.464(5) Å and 2.424(0) Å, respectively. The dihedral angles between the pyridine ring and phenyl groups C4–C5–C6–C7–C8–C9 (phenyl 1) and C17–C18–C19–C20–C21–C22 (phenyl 2) are 32.43(4)° and 25.46(4)°, respectively. It is worth noting that the two intramolecular three-center H-bonds are twisted and not in the same plane due to the steric crowding between the two adjacent propoxy groups. As in 1a, the two amide nitrogens in 3a are also sp2 hybridized for the bond angles across each of the two amide nitrogens N1 and N3 are ca. 120° and the mean deviations of the two amide group C9–N1–H1–C10–O2–C11 (plane 1) and C15–C16–O3–N3–H3–C17 (plane 2) are 0.0341 and 0.0417, respectively. The dihedral angles between the pyridine ring and the amide groups plane 1 and plane 2 are 5.39(1)° and 2.46(3)°, respectively.
| D | H | A | d(H⋯A)/Å | ∠D–H⋯A/° |
|---|---|---|---|---|
| N1 | H1 | N2 | 2.219(7) | 111.59(1) |
| N1 | H1 | O1 | 2.277(6) | 101.14(1) |
| N3 | H3 | N2 | 2.223(5) | 111.30(9) |
| N3 | H3 | O4 | 2.254(9) | 103.11(2) |
| C8 | H8 | O2 | 2.464(5) | 112.99(0) |
| C18 | H18 | O3 | 2.424(0) | 114.78(2) |
A similar compound with methoxy groups, N,N′-bis(2-methoxyphenyl)pyridine-2,6-dicarboxamide, gave a crystal structure analogous to 3a where two three-center H-bonds were also observed.29a
Intramolecular hydrogen bonds (H-bonds) in solution
Infrared spectrum could only provide evidence of hydrogen bond of 3 in CHCl3 (see ESI, Fig. S24 and S26†), but it is impossible to distinguish intramolecular from intermolecular hydrogen bonding interactions. The bands due to hydrogen bonded NH stretching of 3a and 3b were found to shift towards lower wavenumber of respective 3372 and 3397 cm−1 compared to higher wavenumber of more than 3400 cm−1 of common free amide NH.30a Thus, to verify the presence of intramolecular H-bonds in picolinamides 3 without N-substitution, the temperature coefficients dδH/dT of N–H were determined in the temperature range between 298 K to 333 K (in steps of 5 K) in CDCl3 or DMSO-d6/CDCl3 (2/8, v/v) by variable-temperature 1H NMR experiments (Fig. 2). It is generally accepted that in nonpolar solvents when the coefficient is less negative than −3 ppb K−1, the hydrogen bonding interaction is considered as intramolecular; when it is more negative than −5 ppb K−1, it is taken as intermolecular hydrogen bond.30 The dδH/dT of N–H in 3a was measured to be −1.58 ppb K−1 in DMSO-d6/CDCl3 (2/8, v/v), which shows a small variation with temperature. This suggests the high possibility of the presence of intramolecular H-bonds even in a polar solvent.31 Therefore, these results are in accord with the observation of presence of intramolecular H-bonds in the crystal structure of 3a. For 3b, the dδH/dT of N–H was measured to be −1.62 ppb K−1 in nonpolar CDCl3 and −5.71 ppb K−1 in polar solvent DMSO-d6/CDCl3 (2/8, v/v). The small variation of dδH/dT in nonpolar solvent also discloses the presence of intramolecular H-bonds in 3b. Both of the infrared spectra and temperature coefficients data indicate that the two-center hydrogen bonds in 3b are less stable than the three-center hydrogen bonds in 3a.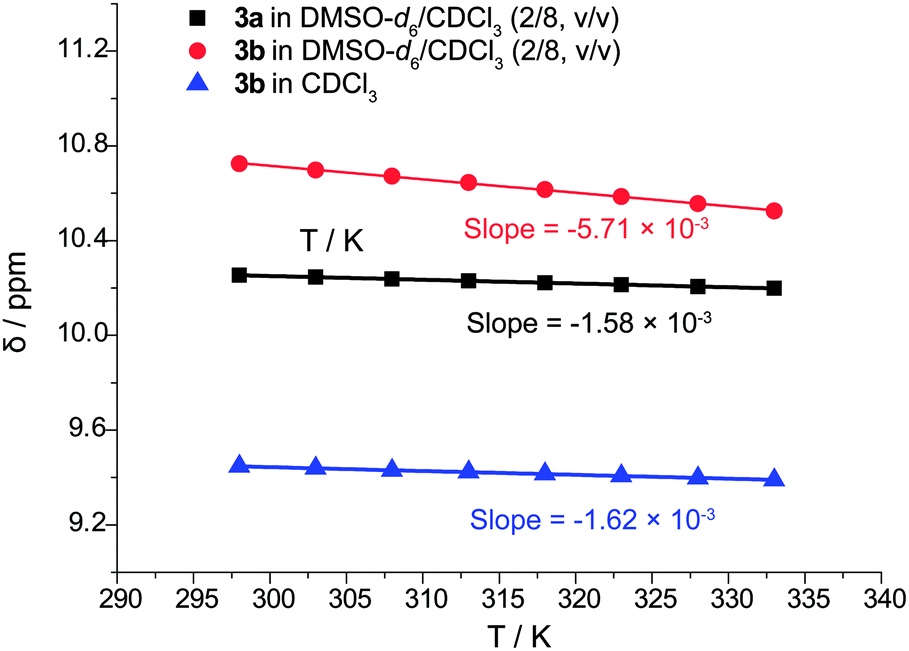 | ||
| Fig. 2 Chemical shifts of NH in 3a/3b versus temperature in DMSO-d6/CDCl3 (2/8, v/v) or CDCl3 (600 MHz, 298 K to 333 K). | ||
Rotational isomerization
Comparison of the 1H NMR spectra of compounds 1a–1c and 3a or 3b disclosed a significant difference in complicacy of signal patterns and chemical shifts of protons b and a on the pyridine moiety (Fig. 3). | ||
| Fig. 3 1H NMR spectra of compounds (a) 1a, (b) 1b, (c) 1c, (d) 3a and (e) 3b in CDCl3 (400 MHz, 298 K). Sign “*” represents signals of solvents. | ||
Among the three positional isomers 1a–1c, 1a exhibits the most poorly-resolved signals over the full spectrum (Fig. 3a). We exclude the possibility of contamination of impurities by HPLC and MS detection (see ESI†). Compound 2, a well-known extractant25,26 with N-ethylated substituent, but free of any replacement on benzene ring, showed similar indistinguishable signals (see ESI†). In stark contrast, 1c, which bears a propoxy group at para-position, provided a much “clean” spectrum with distinguishable signals for almost all aromatic and aliphatic protons a ∼ i (Fig. 3c). Apart from the major clear signals, there are some signals of very low intensity, suggestive of the presence of other rotational isomers. For 1b, the situation sits in between 1a and 1c (Fig. 3b). Signals of each proton from various rotamers of 1b show a tendency to coalesce together but are broad. If compared to the clear, well-resolved signals for 3a (Fig. 3d), these observations strongly suggest that the spectral complexity of 1a is more likely to arise from the concurrent rotamers, rotational isomers that result from the hindered rotations about OC–N bonds and N (amide)–C (Ph).23b,32 It should be noted that the molecular skeleton of 3a is preorganized by aid of intramolecular three-center hydrogen bonds to take a crescent conformation. The two localized intramolecular hydrogen bonds each consist of two S(5)-type rings that involve the backbone amide hydrogen. It has been well established that this three-center hydrogen bond is highly stable, the presence of which hinders the rotational freedom of the aromatic amide-based backbones.33 Therefore, the shape-persistency endows the molecule of 3a with drastically-reduced rotation with respect to 1a–1c in solution, leading to an indication of presence of only one species in solution in 1H NMR spectrum. Furthermore, the rigid backbone also renders the preorganized carbonyl oxygen atoms of the molecule point outwards in 3a. Similarly, the presence of two intramolecular hydrogen bonds in 3b also enforces globally curved conformation of the molecular backbone,23b,34 thus hindering the rotation about OC–N amide bond as manifested in its clear proton signals (Fig. 3e).
The difference of 13C NMR spectra (Fig. 4) among 1a–1c, 3a and 3b was also observed. If there is no presence of rotational isomers in 1a–1c, with the molecular formula C29H35N3O4, the number of signals should be equal to or less than 15 due to their structural symmetry. In fact, there are totally 29 strong signals in 13C NMR spectrum of 1a along with some very weak signals across the full spectrum that could be detected (Fig. 4a), suggesting the presence of a mixture of several major and minor rotamers in solution. This is consistent with the observation of several sets of indistinct 1H NMR signals (Fig. 3a). With 1b, a structural isomer of 1a with propoxy groups at meta-position of the benzene ring, the number of signals in 13C NMR spectrum is drastically decreased to only 15, but some of the signals are broad (Fig. 4b). Compound 1c, which bears a para-substituent, gives a spectrum containing only 13 signals along with another set of very weak signals (Fig. 4c). The decreased number of signals from 13C NMR data suggests that the rotational barrier decreases with change of substituents from ortho to meta to para position. This is different from the result from 3a and 3b where only well-resolved 13 and 11 signals were observed (Fig. 4d and e), respectively, corresponding exactly to respective 25 and 23 carbons in the molecules due to their shape-persistency of molecular backbone rigidified by intramolecular hydrogen bonds. These results agree well with those from 1H NMR spectra. Given the fact that the formation of rotamers is caused by the limitation of CO–N bond rotation, reduction of steric hindrance via alternation of substitution position would lead to decreased rotational barrier and thus simple NMR patterns. Indeed, as the steric hindrance between alkoxyphenyl group and N–Et group increases with the substitution position in order of ortho > meta > para, the signals in both 13C and 1H NMR change from complex to simple and well-resolved.
 | ||
| Fig. 4 13C NMR spectra of compounds (a) 1a, (b) 1b, (c) 1c, (d) 3a and (e) 3b in CDCl3 (100 MHz, 298 K). Red and blue arrows represent some of the major and minor rotational isomers, respectively. | ||
To further probe rotational isomerization, variable-temperature NMR spectra were recorded using DMSO-d6 solutions of 1a as a typical example in the temperature range from 298 K to 428 K (in steps of 10 K) for 1H NMR and 298 K to 418 K (in steps of 20 K) for 13C NMR. In 1H NMR spectra (Fig. 5), each proton of 1a presents complicated multiple sets of signals resulting from various rotamers in solution at 298 K. Owning to the overlapping of signals, it is difficult to identify the species and calculate equilibrium ratio for the rotamer mixture. With the increase of temperature, all the signals in 1a coalesce from complex to broad into a set of distinguishable signals at approximate 408 K. In 13C NMR spectra (Fig. 6), the number of the signals of 1a decreased from 29 at 298 K to 15 at about 398 K due to coalescence effect. Based on the overall change from both 1H NMR and 13C NMR, the coalescence temperature for 1a is reasonably set at approximately 408 K. It is not possible to calculate the temperature coefficients and the free energy, enthalpy and entropy of activation for the interconversion between each rotamers by Eyring analysis because of the complexity of 1H NMR in 1a.
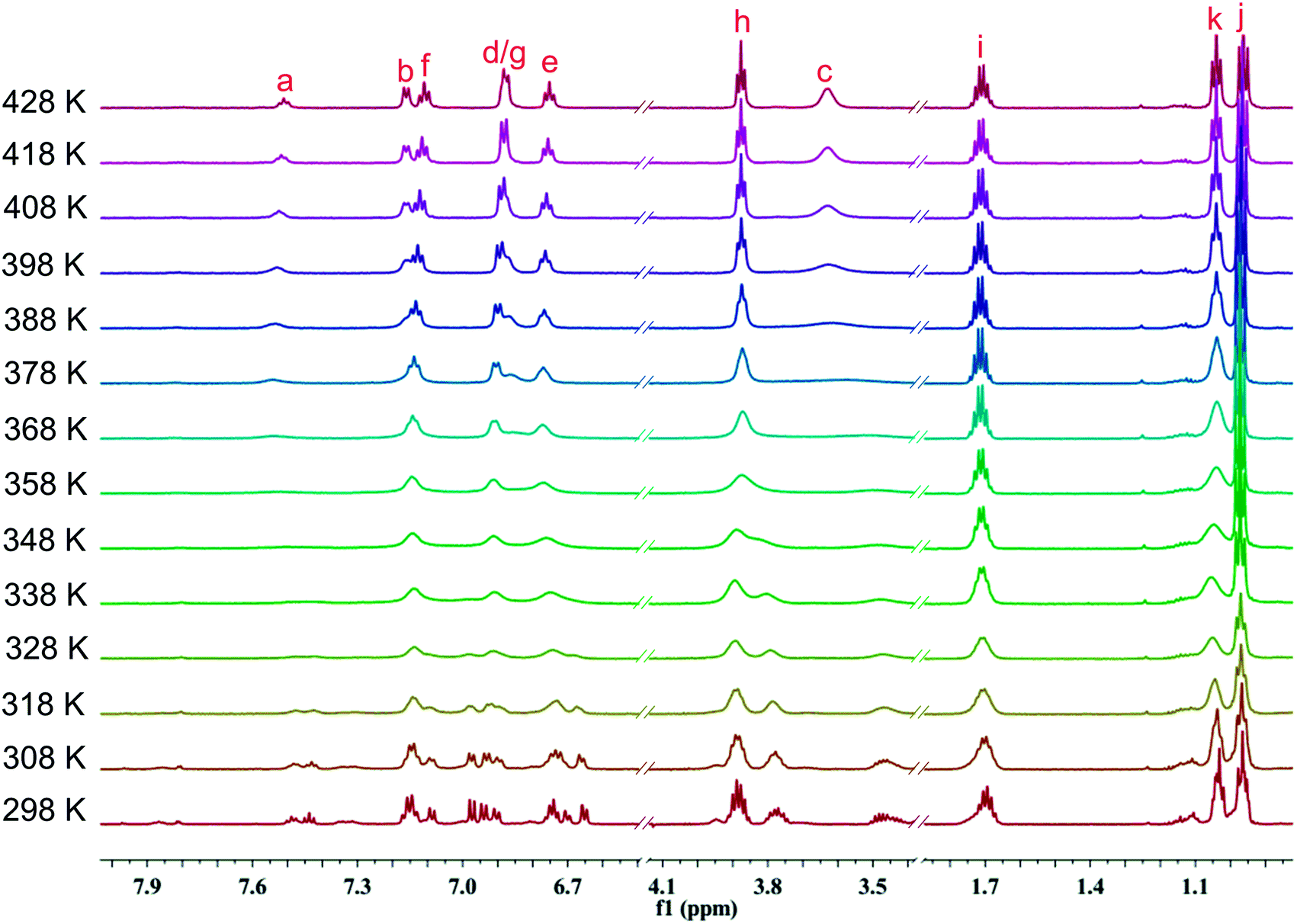 | ||
| Fig. 5 Temperature dependent 1H NMR spectra of 1a in DMSO-d6 in the range from 298 K to 428 K (600 MHz). | ||
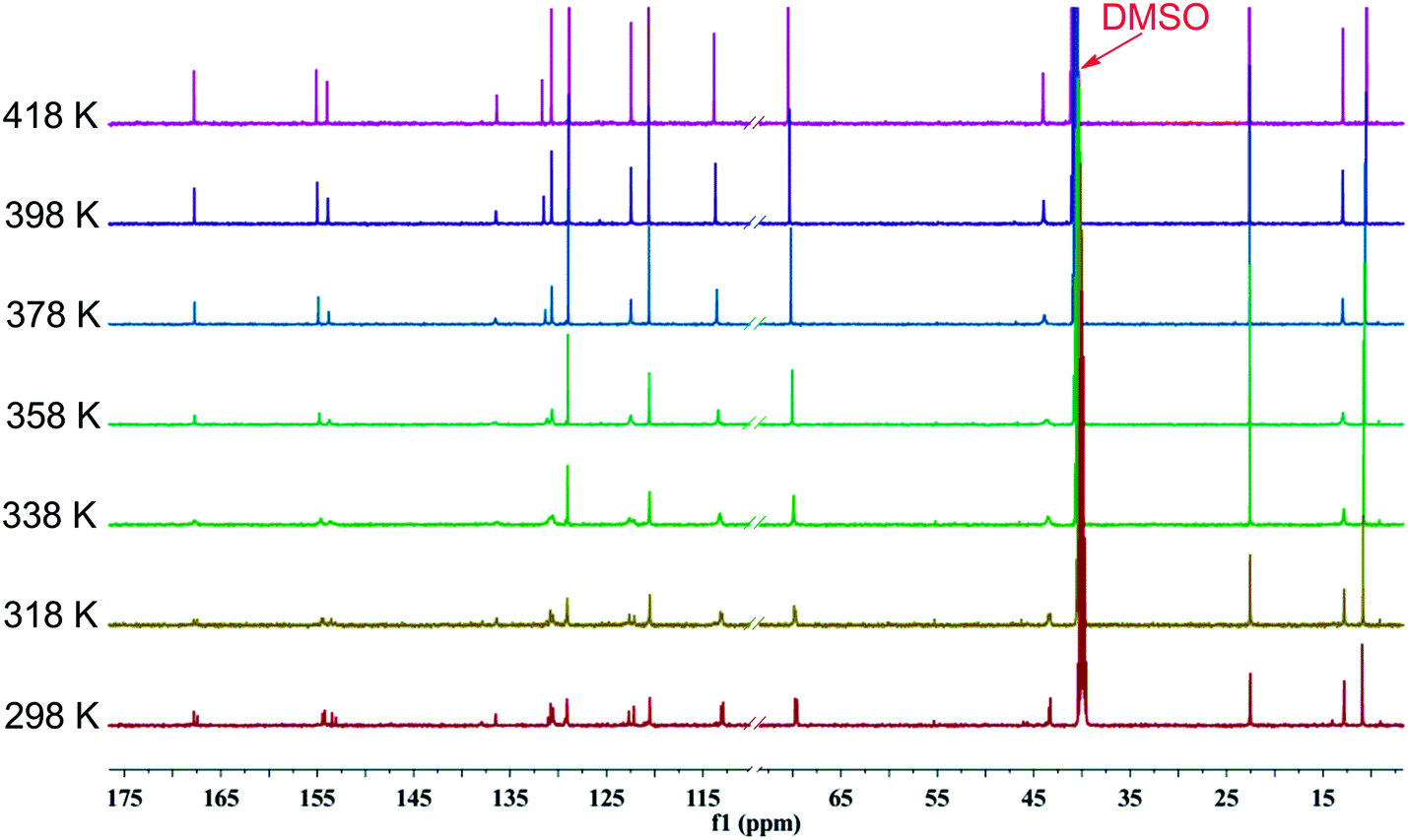 | ||
| Fig. 6 Temperature dependent 13C NMR spectra of 1a in DMSO-d6 in the range from 298 K to 418 K (150 MHz). | ||
To clearly describe rotational isomerization, conformation designation is denoted by Z, E, syn and anti, respectively (vide supra). For 1a, there should exist about ten typical rotational isomers theoretically due to the blocked rotations about the OC–N amide bonds and OC–C (pyridine) bonds (Scheme 2a), which explains undistinguished signals on the NMR timescale. The molecular structure of 1a in the solid state confirms the exclusive formation of the E–anti–anti–E isomer. The two amide nitrogens are in anti conformation with respect to pyridine nitrogen. This is quite different from the solid state structure of the previously reported analogue 2 (ref. 11a) or 9 (ref. 35) bearing no substituents where the E–anti–syn–E conformation was observed (Scheme 2d). In both cases of 1a and 2 (or 9), the E conformation as designated around amide bond is mainly attributed to the outcome of n–π repulsion, i.e., electronic repulsion between the electron-dense center of the amide oxygen and the phenyl ring.28c,36 Computer modeling37 disclosed the higher energy for anti–Z conformation among four possible combinations: anti–Z, anti–E, syn–Z, and syn–E, from which only six reasonable conformations E–syn–syn–E, E–anti–syn–E, E–anti–anti–E, E–anti–syn–Z, E–syn–syn–Z, Z–syn–syn–Z were obtained (Fig. 7). In the anti–Z conformation, carbonyl oxygen atoms experience both n–n repulsion with the pyridine nitrogen and n–π repulsion with the phenyl ring, leading to much lower stability of the conformation. Thus, the observed E–anti–anti–E conformation (1a) in the solid state is consistent to one of the calculated results.
 | ||
| Fig. 7 Optimized rotational structures of compound 1a obtained by DFT calculation at the B3LYP/6-31G(d) level. | ||
Liquid–liquid extraction
The structural difference between 1a and 3a is indeed manifested in the following extractive results.Eight transition metal picrates were employed in liquid–liquid extraction experiments including picrate salts of Ag+, Hg2+, Pb2+, Cd2+, Zn2+, Cu2+, Co2+ and Ni2+. The extraction abilities of picolinamides 1a–1c, 2, 3a and 3b towards these metal ions were examined by the standard picrate extraction method.38 Compound 2 bearing no substituent on benzene rings was employed as a control for 1a–1c.
Results from extraction of the above transition metal ions from water into dichloromethane are shown in Table 3 and Fig. 8. All ligands exhibited good to excellent extraction ability for Hg2+. Almost no extraction or small extraction (<9%) was detected for Pb2+, Cd2+, Zn2+, Co2+ and Ni2+ except for 1a for Cu2+. Ligand 1a extracted almost exclusively Hg2+ compared to other ions. Particularly noteworthy is the remarkable difference in extraction of Hg2+ with ligand 3a containing intramolecular H-bonds and its N-substituted analogues 1a–1c. For example, ligand 1a showed extractability of 95.4% for Hg2+, while 3a gave a lower value of 38.4%. The large difference (57.0%) for 1a as compared to 3a is also revealed in 1b and 1c, which enhanced the extraction by 50.2% and 54.6%, respectively. Compound 2, also ethylated on amide nitrogens, behaved in a similar fashion and showed a relatively large difference of 38.6% in extracting Hg2+ compared to 3a. In principle, effective coordination of the ligand with metal ions requires the orientation of two carbonyl oxygen atoms and nitrogen of the pyridine moiety to be arrayed on the same side.9d,39 For 3a, orientation of carbonyl oxygens inwards in line with the pyridine nitrogen is impossible due to rigidified backbone by intramolecular hydrogen bonds. However, driven by the presence of metal ions transferred from the aqueous phase in the course of extraction, rotation of carbonyl oxygens to direct right coordination is more likely to occur for 1a–1c even for rotamers of high energy (e.g., E–syn–syn–Z, Z–syn–syn–Z) since the intramolecular H-bonding was disrupted in 1a–1c after ethylation of amide nitrogen. This explains the higher extractability of Hg2+ for these compounds (88.6–95.4%) compared to 3a (38.4%). In fact, use of another compound 3b containing two intramolecular hydrogen bonds afforded an extractability of 40.6% for Hg2+, which is very close to that with 3a as extractant. This indicates that intramolecularly hydrogen-bonded extractants (3a and 3b) are inferior to those containing no hydrogen bonds, underscoring the importance of released constraint of rotational restriction for chelating metal ions upon extraction.
| Metal ion | Hydration energy41 ΔGhyd (kJ mol−1) | Extractionb (%) | |||||
|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 2 | 3a | 3b | ||
| a Aqueous phase (10 mL); [Pic−] = 2 × 10−5 M, organic phase (10 mL); [L] = 2 × 10−4 M, 298 K.b Average for three independent extraction experiments. | |||||||
| Hg2+ | −1760 | 95.4 ± 0.4 | 88.6 ± 1.0 | 93.0 ± 0.4 | 77.0 ± 0.5 | 38.4 ± 0.4 | 40.6 ± 0.2 |
| Ag+ | −430 | 58.7 ± 0.6 | 38.5 ± 0 | 42.1 ± 0.2 | 27.4 ± 0.4 | 2.9 ± 0.2 | 3.2 ± 0.2 |
| Cu2+ | −2010 | 31.5 ± 0.4 | 1.9 ± 0.2 | 2.1 ± 0.4 | 3.3 ± 0.7 | 1.5 ± 0.2 | 3.0 ± 0.7 |
| Ni2+ | −1980 | 8.2 ± 0.2 | 1.5 ± 0.2 | 0.5 ± 0.5 | 2.6 ± 0.5 | 0.9 ± 0.2 | 2.5 ± 0.2 |
| Cd2+ | −1755 | 3.0 ± 0.5 | 0.8 ± 0.9 | 0.6 ± 0.5 | 3.1 ± 0.3 | 0.7 ± 0 | 1.2 ± 0.2 |
| Co2+ | −1915 | 4.9 ± 0 | 0.8 ± 0.9 | 0.9 ± 0.2 | 0.2 ± 0.4 | 1.2 ± 0.2 | 3.3 ± 0.7 |
| Zn2+ | −1955 | 7.2 ± 0.5 | 0.3 ± 0.5 | 0.6 ± 0.2 | 1.9 ± 0.7 | 2.2 ± 0.2 | 1.5 ± 0.7 |
| Pb2+ | −1425 | 7.9 ± 0.5 | 4.4 ± 0.2 | 3.6 ± 0.2 | 3.5 ± 0.5 | 1.2 ± 0.2 | 3.2 ± 0.5 |
 | ||
| Fig. 8 Extraction of transition metal picrates by compounds 1a–1c, 2, 3a and 3b from water into dichloromethane at 298 K. Aqueous phase (10 mL); [Pic−] = 2 × 10−5 M, organic phase (10 mL); [L] = 2 × 10−4 M. | ||
On the other hand, N-substituted groups can increase the basicity, nucleophilicity and softness of the coordinating amide groups and the lipophilicity of the extracted complex compared to N–H groups, which would also be one of reasons to enhance the extractability of 1a–1c and 2.40 As shown in Table 3, the extraction percentage decreased in the order of Hg2+ > Ag+ > Cu2+ > (or ∼) Ni2+ ∼ Co2+ ∼ Zn2+ ∼ Pb2+ ∼ Cd2+, which does not follow the order of hydration energy41 or ionic radii of the metal ions41 (Pb2+ > Ag+ > Hg2+ > Cd2+ > Zn2+ ∼ Co2+ > Cu2+ > Ni2+), suggesting that the higher selectivity towards Hg2+could be attributed to the synergy of several factors such as hydration of metal ions, ionic radius, charge number and hardness/softness42 between the nitrogen-containing ligands and Hg2+. The effect of structural difference as in 1a–1c, 2, 3a and 3b upon extraction behaviour was also unraveled by the results from extraction of Ag+. Ligand 1a, 1b, 1c, and 2 extracted Ag+ in 58.7%, 38.5%, 42.1% and 27.4%, respectively; however, the extractability for 3a and 3b is very low (<4%), again demonstrating the dependence of extraction upon the presence of intramolecular hydrogen bonds.
Regarding the extraction difference among 1a, 1b and 1c and 2, electronic effect seems to play a major role, which arises from different substitution position of propoxy groups on benzene rings. Ortho- and para-substitution provided highest extraction results (95.4% and 93.0%). The efficiency decreased by ca. 17% for compound 2 having no electron-donating groups. Among the four ligands 1a–1c and 2, 1c is not only with high extractability but also much more selective towards extracting Hg2+ than other metal cations.
To comprehend the complexing behaviour of extracted species in the extraction process, the stoichiometries of the ligands and metal cations were measured. The dependence of log{D/[Pic−]n} as a function of the concentration of ligands 1a–1c at constant Hg-picrate concentration offers a linear relationship between log{D/[Pic−]n} and log[L] with the slopes of 2.26, 1.96 and 2.15 for 1a, 1b and 1c, respectively (Fig. 9). This implicates the presence of the extracted species in approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (L:M) between 1a–1c and Hg2+. The values of the extraction constants logKex were calculated to be 17.07, 15.47 and 16.62 for 1a, 1b and 1c, respectively.
:1 (L:M) between 1a–1c and Hg2+. The values of the extraction constants logKex were calculated to be 17.07, 15.47 and 16.62 for 1a, 1b and 1c, respectively.
 | ||
| Fig. 9 Plot of log{D/[Pic−]n} versus log[L] for the extraction of Hg-picrate with ligands 1a–1c. | ||
Furthermore, the method of Job's plot was used to supply more information on the Hg2+ binding stoichiometry of 1a–1c. The resulting Job's plot of 1a/1b/1c–Hg2+ complexation is shown in Fig. 10. The maximum absorbance is observed at 0.67, indicating a ligand–metal ratio of 2:1 in the complex. On the basis of 2:1 stoichiometry and UV-vis titration data (see ESI, Fig. S29–31†), the binding constants K1 and K2 of 1a–Hg2+ in CH3CN are estimated to be 3.34 × 107 M−1 and 1.38 × 106 M−1 (Fig. 11) using nonlinear curve fitting method.43 Similarly, the binding constants of 1b–Hg2+ and 1c–Hg2+ are also estimated using the same method (see ESI, Fig. S32 and 33†).
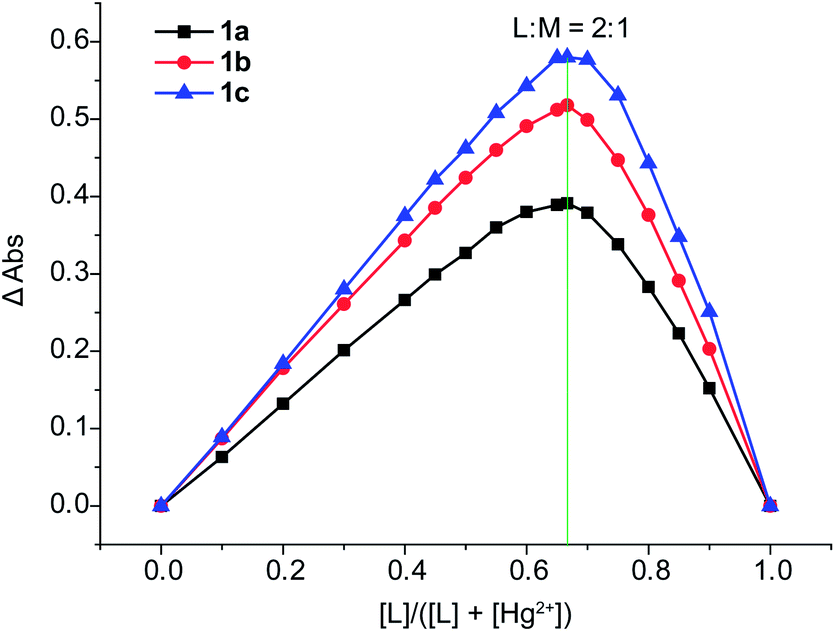 | ||
| Fig. 10 Job's plot for the determination of stoichiometry in the complex formed by 1a–1c and Hg2+ from absorbance measurements in CH3CN. | ||
 | ||
| Fig. 11 Curve-fitting analysis for the complexation of 1a with Hg2+ in CH3CN. | ||
To understand the coordinate sites of the ligands, the complex 1a–Hg2+ was prepared from a CH3CN solution containing 1a and Hg(NO3)2 in molar ratio 2:1 and its infrared spectrum was compared to that of the free ligand 1a (Fig. 12). The strong band at 1649 cm−1 of ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in 1a shifts to 1632 cm−1 in the complex, a change of 17 cm−1 from vibration of carbonyl oxygen, indicative of the involvement of oxygen atoms in coordination. Since the ν(OC–N) band at 1264 cm−1 for amide bonds in 1a only shifts upward by 2 cm−1 upon complexation, it suggests that the two amide nitrogens are not involved in the coordination with metal ions. Besides, the band of pyridine ring vibrations appears at 1475 cm−1 in free 1a and merges into a band at 1456 cm−1 in coordinated 1a. Based on these observations, we conclude that the coordinate atoms of 1a should come from carbonyl oxygens, and pyridine nitrogen is also involved.
O) in 1a shifts to 1632 cm−1 in the complex, a change of 17 cm−1 from vibration of carbonyl oxygen, indicative of the involvement of oxygen atoms in coordination. Since the ν(OC–N) band at 1264 cm−1 for amide bonds in 1a only shifts upward by 2 cm−1 upon complexation, it suggests that the two amide nitrogens are not involved in the coordination with metal ions. Besides, the band of pyridine ring vibrations appears at 1475 cm−1 in free 1a and merges into a band at 1456 cm−1 in coordinated 1a. Based on these observations, we conclude that the coordinate atoms of 1a should come from carbonyl oxygens, and pyridine nitrogen is also involved.
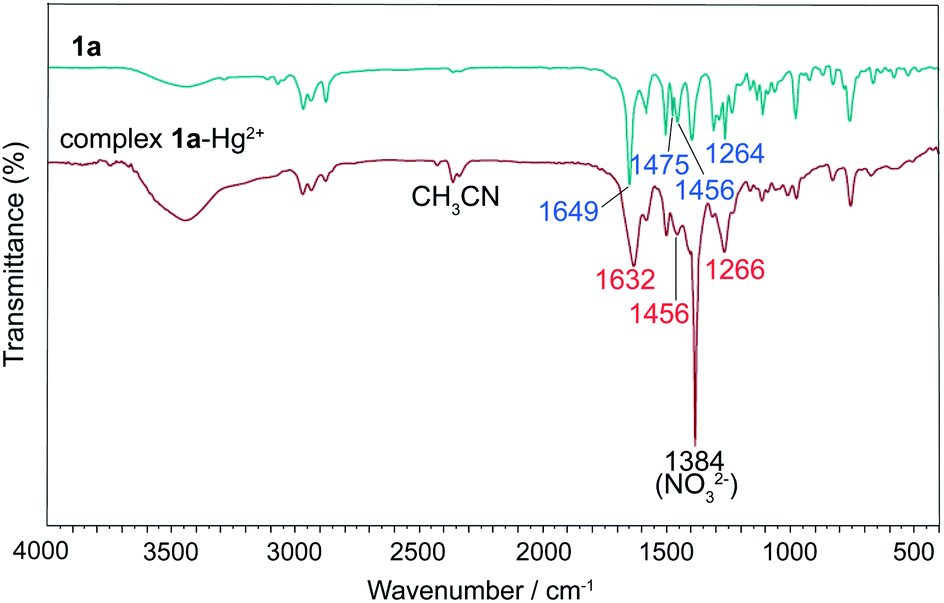 | ||
| Fig. 12 Infrared spectra of 1a and the complex 1a–Hg2+. | ||
The complexation of 1a–1c, 3a and 3b with Hg2+ were evidenced by the spectral change in the 1H NMR experiments (Fig. 13 and 14). In CD3CN/CDCl3 (1/9, v/v), almost all of the protons experience a downfield shift for compound 1a–1c upon addition of Hg2+. In sharp contrast, for compounds 3a and 3b, neither chemical shifts nor signal patterns undergo any change, strongly suggesting that the interaction of 1a–1c with Hg2+ is much stronger than that of 3a or 3b. This explains the much higher efficiency as indicated for 1a–1c as extractants compared to 3a and 3b. In general, the NMR patterns of 1a–1c tend to become simple after addition of Hg2+. In the case of 1a, upon complexing the metal ion, though still poorly-resolved, the signal pattern (Fig. 13b) resembles that from variable-temperature 1H NMR experiments (Fig. 5 at 318 K). For 1b, the broadened signals change to one set of well-resolved sharp signals in the presence of Hg2+ (Fig. 13c and d), suggesting the transformation from mixed multiple rotational isomers to only one major isomer induced by introduction of metal ion. The similar result was obtained for 1c, where minor signals of very low intensity and major signals merge into one set of broad signals at aromatic region (Fig. 13e and f, 6.5–7.6 ppm), while aliphatic protons (0.8–4.2 ppm) become more distinguishable. These results suggest that complexation of Hg2+ by 1a–1c facilitate the reduction of possible rotational isomers in solution, but have no influence upon isomerism for intramolecularly hydrogen-bonded compounds 3a and 3b, again underscoring the importance of hydrogen bonding and rotational isomerism on extraction.
 | ||
| Fig. 13 Partial 1H NMR spectra in 10% CD3CN/90% CDCl3 (400 MHz, 298 K): (a) 1a; (b) 1a + Hg(NO3)2 (2:1); (c) 1b; (d) 1b + Hg(NO3)2 (2:1); (e) 1c; (f) 1c + Hg(NO3)2 (2:1). Sign “*” represents signals of solvents. | ||
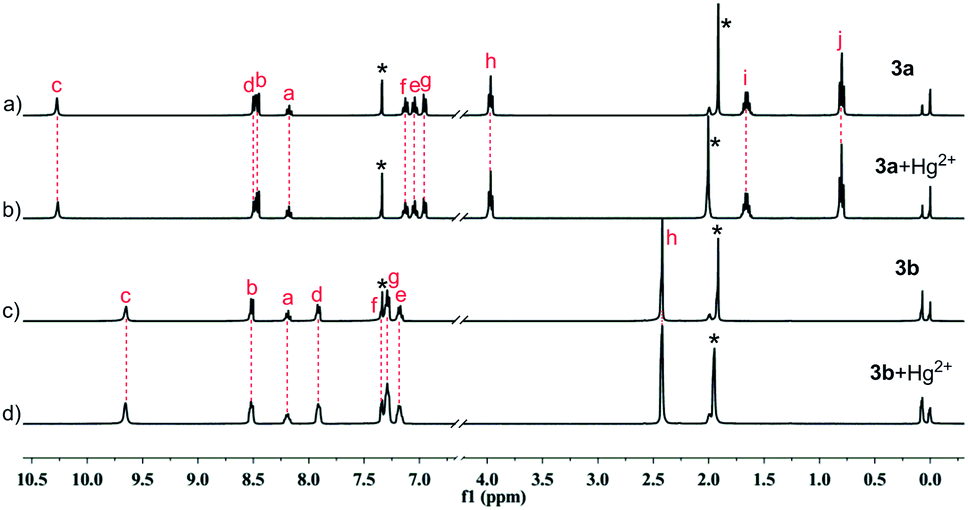 | ||
| Fig. 14 Partial 1H NMR spectra in 10% CD3CN/90% CDCl3 (400 MHz, 298 K): (a) 3a; (b) 3a + Hg(NO3)2 (2:1); (c) 3b; (d) 3b + Hg(NO3)2 (2:1). Sign “*” represents signals of solvents. | ||
Experimental
Materials and reagents
Compounds 4 and 5a–5c were synthesized following the similar reported procedures.44,45 Compound 7 was prepared from hydrogenation of 5a in almost quantitative yield (see ESI†). Dichloromethane, picric acid, anhydrous Na2SO4, Hg(NO3)2·H2O, AgNO3, Cu(NO3)2·3H2O, Ni(NO3)2·6H2O, Cd(NO3)2·4H2O, Zn(NO3)2·6H2O, Co(NO3)2·6H2O, Pb(NO3)2 were the analytical grade reagents and were purchased from Chengdu Kelong Chemical Factory. All other solvents and chemicals used for the synthesis were of reagent grade and used as received.Instruments and apparatus
UV-vis spectra were measured by SHIMADZU UV-2350. 1H NMR and 13C spectra were recorded on Bruker AVANCE AV II-400 MHz (1H: 400 MHz; 13C: 100 MHz). Chemical shifts are reported in δ values in ppm and coupling constants (J) are denoted in Hz. Multiplicities are denoted as follows: s = singlet, d = doublet, t = triplet, and m = multiplet. High resolution mass data were collected by WATERS Q-TOF Premier. CDCl3, DMSO-d6 and CD3CN were from Cambridge Isotope Laboratories (CIL).Synthesis of compound 6a–6c
Compound 6a–6c was synthesized following the reported procedure in a yield of 79%, 81%, 73%, respectively.27 After two vacuum/H2 cycles to remove air from the reaction, the stirred mixture of the nitropropoxybenzene 5a/5b/5c (1.00 g, 5.52 mmol), 100 mg 10% Pd/C and 50 mL acetonitrile was hydrogenated at ordinary pressure and at room temperature. The reaction was monitored using TLC until the secondary amine was no longer increased. The reaction mixture was filtrated and the filtrate was concentrated under reduced pressure. The crude mixture was purified by flash silica gel column chromatography, and provided the product as light yellow oil, which was used for the immediate coupling reaction.Synthesis of pyridine-based 2,6-dicarboxyamides 1a–1c, 3a and 3b
The general procedure for compounds 1a–1c, 3a and 3b was exemplified by the synthesis of 1a. Triethylamine (3.35 g, 33.12 mmol) was added into a solution of the amine 6a (3.32 g, 22.0 mmol) in 100 mL of dry dichloromethane at 0 °C under N2. Pyridine-2,6-dicarbonyl dichloride 4 (2.24 g, 11.0 mmol) was dissolved in 50 mL of dichloromethane and added dropwise to the above mixture. The solution was stirred at room temperature under N2 for 4 h. The organic layer was washed with 10% HCl aqueous and followed water, and dried over anhydrous Na2SO4 and filtered. Most volatiles were removed under reduced pressure and the residue was isolated by precipitation by addition of methanol to give a white solid.Solvent extraction
Heavy metal picrates were prepared by the stepwise addition of 1 × 10−2 M of metal nitrate solution to 2 × 10−5 M aqueous picric acid solution and shaken at 298 K for 1 h. 10 mL of a 2 × 10−5 M aqueous metal picrate solution and 10 mL of a 2 × 10−4 M solution of ligands in CH2Cl2 were placed in a stoppered glass tube and vigorously agitated with a mechanical shaker in a thermostated water bath at 298 K for 2 h. The resulting mixtures were left standing for an additional 2 h in order to complete the phase separation. The concentration of the picrate anion remaining in the aqueous phase was determined by UV spectrophotometry at λmax 355 nm. Blank experiments showed that no picrate extraction occurred in the absence of ligands. The extractability was determined based on the absorbance of picrate anion in the aqueous solutions. The extractability (E%) was calculated based on the equation: E% = 100(A0 − A)/A0, where A0 is the absorbance of the aqueous solution in the absence of ligand, A is the absorbance of the aqueous phase after extraction. Three independent experiments were carried out and the average value of percent picrate extracted was calculated.Conclusion
In summary, a pyridine-based aromatic amides 1a–1c with N-substitution and their analogues 3a and 3b containing intramolecular hydrogen bonds were synthesized for probing the interplay of molecular structure and liquid–liquid extraction behaviour towards transition metal ions. X-ray diffraction analysis of ligands 1a and 3a provides information of molecular conformation without and with intramolecular H-bonding. The observed E–anti–anti–E conformation (1a) in the solid sate is among one of six reasonable rotational isomeric structures of 1a optimized by computer modeling. Ordinary and variable-temperature proton and carbon NMR experiments of 1a–1c disclosed the formation of rotamers due to N-substitution. The fact that N-substitution is responsible for the higher selectivity and efficiency towards Hg2+ over other metal cations is rationalized by the large difference in rotational restriction between N-substituted 1 (a, b and c) and intramolecularly hydrogen bonded 3 (a, b). The results from 1H NMR spectra regarding the interaction of the ligands with Hg2+ also verify the extraction difference, and simultaneously disclose the influence of complexation on rotational isomerism of ligands. Despite the absence of intramolecular hydrogen bonding as compounds 1 (a, b and c), compound 2 still displayed a lower extraction ability than 1 (a, b and c) due to the favorable electronic effect arising from alkoxy substitution. The stoichiometry for the complexation of Hg2+ by 1a–1c was found to be 2:1 (ligand/metal ion) using log{D/[Pic−]n} − log[L] analysis, Job's plot and UV-vis titration. IR study indicates that the coordinate atoms are carbonyl oxygens and pyridine nitrogen in N-substituted ligands. The disclosure of the impact of H-bonding-enforced backbone rigidification and structural variation via N-substitution upon extraction as presented in this work may provide in-depth understanding of the extraction process associated with intramolecular hydrogen bonding and rotational conformation.
Acknowledgements
This work is supported by the National Natural Science Foundation (21172158), NSAF(11076018), the Doctoral Program of the Ministry of Education of China (20130181110023), the National Science Foundation for Fostering Talents in Basic Research of the National Natural Science Foundation of China (J1210004 and J1103315), Open Project of State Key Laboratory of Supramolecular Structure and Materials (SKLSSM201408) and Open Project of State Key Laboratory of Structural Chemistry (20140013). Comprehensive Training Platform of Specialized Laboratory, College of Chemistry, Sichuan University is acknowledged for NMR, HRESI-MS and IR analyses.Notes and references
- D. W. Zhang, X. Zhao, J. L. Hou and Z. T. Li, Chem. Rev., 2012, 112, 5271 CrossRef CAS PubMed.
- (a) A. E. V. Gorden, M. A. DeVore, II and B. A. Maynard, Inorg. Chem., 2013, 52, 3445 CrossRef CAS PubMed; (b) B. J. Mincher, G. Modolo and S. P. Mezyk, Solvent Extr. Ion Exch., 2009, 27, 579 CrossRef CAS.
- (a) H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465 RSC; (b) I. G. Spiridonov, D. O. Kirsanov, V. A. Babain, M. Y. Alyapyshev, N. I. Eliseev, Y. G. Vlasov and A. V. Legin, Russ. J. Appl. Chem., 2011, 84, 1354 CrossRef CAS; (c) Ü. Ocak, M. Ocak, X. Shen, K. Surowiec and R. A. Bartsch, J. Fluoresc., 2009, 19, 997 CrossRef PubMed; (d) Ü. Ocak, M. Ocak, K. Surowiec, X. Liu and R. A. Bartsch, Tetrahedron, 2009, 65, 7038 CrossRef PubMed; (e) M. A. Qazi, Ü. Ocak, M. Ocak, S. Memon and I. B. Solangi, J. Fluoresc., 2013, 23, 575 CrossRef CAS PubMed.
- (a) P. Geoghegan and P. O'Leary, ACS Catal., 2012, 2, 573 CrossRef CAS; (b) N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2013, 52, 223 CrossRef CAS PubMed.
- S. A. Ansari, P. Pathak, P. K. Mohapatra and V. K. Manchanda, Chem. Rev., 2012, 112, 1751 CrossRef CAS PubMed.
- (a) A. Casnati, N. Della Ca', M. Fontanella, F. Sansone, F. Ugozzoli, R. Ungaro, K. Liger and J. F. Dozol, Eur. J. Org. Chem., 2005, 2338 CrossRef CAS; (b) E. Macerata, F. Sansone, L. Baldini, F. Ugozzoli, F. Brisach, J. Haddaoui, V. Hubscher-Bruder, F. Arnaud-Neu, M. Mariani, R. Ungaro and A. Casnati, Eur. J. Org. Chem., 2010, 2675 CrossRef CAS; (c) M. Galletta, L. Baldini, F. Sansone, F. Ugozzoli, R. Ungaro, A. Casnati and M. Mariani, Dalton Trans., 2010, 39, 2546 RSC.
- (a) M. M. Reinoso-García, D. Jańczewski, D. N. Reinhoudt, W. Verboom, E. Malinowska, M. Pietrzak, C. Hill, J. Báča, B. Grüner, P. Selucky and C. Grüttner, New J. Chem., 2006, 30, 1480 RSC; (b) K. Matloka, A. K. Sah, M. W. Peters, P. Srinivasan, A. V. Gelis, M. Regalbuto and M. J. Scott, Inorg. Chem., 2007, 46, 10549 CrossRef CAS PubMed; (c) E. V. Sharova, O. I. Artyushin, A. N. Turanov, V. K. Karandashev, S. B. Meshkova, Z. M. Topilova and I. L. Odinets, Cent. Eur. J. Chem., 2012, 10, 146 CrossRef CAS PubMed.
- (a) A. Shimada, T. Yaita, H. Narita, S. Tachimori and K. Okuno, Solvent Extr. Ion Exch., 2004, 22, 147 CrossRef CAS PubMed; (b) M. Y. Alyapyshev, V. A. Babain, N. E. Borisova, R. N. Kiseleva, D. V. Safronov and M. D. Reshetova, Mendeleev Commun., 2008, 18, 336 CrossRef CAS PubMed; (c) A. Paulenova, M. Y. Alyapyshev, V. A. Babain, R. S. Herbst and J. D. Law, Sep. Sci. Technol., 2008, 43, 2606 CrossRef CAS; (d) M. Y. Alyapyshev, V. A. Babain, L. I. Tkachenko, A. Paulenova, A. A. Popova and N. E. Borisova, Solvent Extr. Ion Exch., 2014, 32, 138 CrossRef CAS.
- (a) J. C. Noveron, M. M. Olmstead and P. K. Mascharak, J. Am. Chem. Soc., 2001, 123, 3247 CrossRef CAS PubMed; (b) T. C. Harrop, L. A. Tyler, M. M. Olmstead and P. K. Mascharak, Eur. J. Inorg. Chem., 2003, 475 CrossRef CAS; (c) R. Kapoor, A. Kataria, A. Pathak, P. Venugopalan, G. Hundal and P. Kapoor, Polyhedron, 2005, 24, 1221 CrossRef CAS PubMed; (d) R. Kapoor, A. Kataria, P. Kapoor and P. Venugopalan, Transition Met. Chem., 2004, 29, 425 CrossRef CAS; (e) R. Kapoor, A. Kataria, P. Venugopalan and P. Kapoor, Inorg. Chem., 2004, 43, 6699 CrossRef CAS PubMed.
- (a) D. O. Kirsanov, N. E. Borisova, M. D. Reshetova, A. V. Ivanov, L. A. Korotkov, I. I. Eliseev, M. Y. Alyapyshev, I. G. Spiridonov, A. V. Legin, Y. G. Vlasov and V. A. Babain, Russ. Chem. Bull., Int. Ed., 2012, 61, 881 CrossRef CAS PubMed; (b) J. L. Lapka, A. Paulenova, M. Y. Alyapyshev, V. A. Babain, R. S. Herbst and J. D. Law, J. Radioanal. Nucl. Chem., 2009, 280, 307 CrossRef CAS PubMed.
- (a) A. Fujiwara, Y. Nakano, T. Yaita and K. Okuno, J. Alloys Compd., 2008, 456, 429 CrossRef CAS PubMed; (b) J. L. Lapka, A. Paulenova, L. N. Zakharov, M. Y. Alyapyshev and V. A. Babain, IOP Conf. Ser.: Mater. Sci. Eng., 2010, 9, 012029 CrossRef.
- V. A. Babain, M. Y. Alyapyshev, I. V. Smirnov and A. Y. Shadrin, Radiochemistry, 2006, 48, 369 CrossRef CAS.
- (a) H. Stephan, K. Gloe, J. Beger and P. Mühl, Solvent Extr. Ion Exch., 1991, 9, 435 CrossRef CAS; (b) Y. Sasaki and G. R. Choppin, Anal. Sci., 1996, 12, 225 CrossRef CAS.
- M. Y. Alyapyshev, V. A. Babain, L. I. Tkachenko, I. I. Eliseev, A. V. Didenko and M. L. Petrov, Solvent Extr. Ion Exch., 2011, 29, 619 CrossRef CAS.
- (a) S. L. Li, T. Xiao, C. Lin and L. Wang, Chem. Soc. Rev., 2012, 41, 5950 RSC; (b) L. Yuan, P. Zhang, W. Feng and B. Gong, Curr. Org. Chem., 2011, 15, 1250 CrossRef CAS; (c) P. K. Baruah and S. Khan, RSC Adv., 2013, 3, 21202 RSC; (d) P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205 RSC.
- (a) Z. Shirin, J. Thompson, L. Liable-Sands, G. P. A. Yap, A. L. Rheingold and A. S. Borovik, J. Chem. Soc., Dalton Trans., 2002, 1714 RSC; (b) J. R. Turkington, P. J. Bailey, J. B. Love, A. M. Wilson and P. A. Tasker, Chem. Commun., 2013, 49, 1891 RSC.
- X. Yang, L. Chen, Y. Yang, Y. He, S. Zou, W. Feng, Y. Yang, N. Liu, J. Liao and L. Yuan, J. Hazard. Mater., 2012, 217–218, 171 CrossRef CAS PubMed.
- (a) L. Yang, L. Zhong, K. Yamato, X. Zhang, W. Feng, P. Deng, L. Yuan, X. C. Zeng and B. Gong, New J. Chem., 2009, 33, 729 RSC; (b) W. Feng, K. Yamato, L. Yang, J. S. Ferguson, L. Zhong, S. Zou, L. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2009, 131, 2629 CrossRef CAS PubMed; (c) Y. Yang, W. Feng, J. Hu, S. Zou, R. Gao, K. Yamato, M. Kline, Z. Cai, Y. Gao, Y. Wang, L. Li, Y. Ynag, L. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2011, 133, 18590 CrossRef CAS PubMed; (d) J. Hu, L. Chen, Y. Ren, P. Deng, X. Li, Y. Wang, Y. Jia, J. Luo, X. Yang, W. Feng and L. Yuan, Org. Lett., 2013, 15, 4670 CrossRef CAS PubMed.
- L. Zhong, L. Chen, W. Feng, S. Zou, Y. Yang, N. Liu and L. Yuan, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 367 CrossRef CAS PubMed.
- L. He, Q. Jiang, Y. Jia, Y. Fang, S. Zou, Y. Yang, J. Liao, N. Liu, W. Feng, S. Luo, Y. Yang, L. Yang and L. Yuan, J. Chem. Technol. Biotechnol., 2013, 88, 1930 CrossRef CAS.
- H. Chu, L. He, Q. Jiang, Y. Fang, Y. Jia, X. Yuan, S. Zou, X. Li, W. Feng, Y. Yang, N. Liu, S. Luo, Y. Yang, L. Yang and L. Yuan, J. Hazard. Mater., 2014, 264, 211 CrossRef CAS PubMed.
- (a) J. L Lapka, A. Paulenova, M. Y. Alyapyshev, V. A. Babain, J. D. Law and R. S. Herbst, IOP Conf. Ser.: Mater. Sci. Eng., 2010, 9, 012068 CrossRef; (b) J. L. Lapka, A. Paulenova, R. S. Herbst and J. D. Law, Sep. Sci. Technol., 2010, 45, 1706 CrossRef CAS; (c) M. Y. Alyapyshev, V. A. Babain, L. I. Tkachenko, I. I. Eliseev, A. V. Didenko and M. L. Petrov, Solvent Extr. Ion Exch., 2011, 29, 619 CrossRef CAS; (d) A. Paulenova, M. Y. Alyapyshev, V. A. Babain, R. S. Herbst and J. D. Law, Solvent Extr. Ion Exch., 2013, 31, 184 CrossRef CAS.
- (a) M. Alyapyshev, V. Babain, N. Borisova, I. Eliseev, D. Kirsanov, A. Kostin, A. Legin, M. Reshetova and Z. Smirnova, Polyhedron, 2010, 29, 1998 CrossRef CAS PubMed; (b) T. L. Borgne, J. M. Bénech, S. Floguet, G. Bernardinelli, C. Aliprandini, P. Bettens and C. Piguet, Dalton Trans., 2003, 3856 RSC.
- L. Wu, Y. Fang, Y. Jia, Y. Yang, J. Liao, N. Liu, X. Yang, W. Feng, J. Ming and L. Yuan, Dalton Trans., 2014, 43, 3835 RSC.
- (a) V. A. Babain, M. Y. Alyapyshev and R. N. Kiseleva, Radiochim. Acta, 2007, 95, 217 CrossRef CAS; (b) E. Makrlik, P. Vaňura, P. Selucký, V. A. Babain and M. Y. Alyapyshev, Radiochemistry, 2009, 51, 479 CrossRef CAS; (c) D. O. Kirsanov, O. V. Mednova, E. N. Pol'shin, A. V. Legin, M. Y. Alyapyshev, I. I. Eliseev, V. A. Babain and Y. G. Vlasov, Russ. J. Appl. Chem., 2009, 82, 247 CrossRef CAS; (d) E. Makrlík, P. Vaňura, P. Selucký, V. A. Babain and I. V. Smirnov, J. Radioanal. Nucl. Chem., 2010, 283, 839 CrossRef PubMed; (e) M. Y. Alyapyshev, V. A. Babain, L. I. Tkachenko, I. I. Eliseev, A. V. Didenko and M. L. Petrov, Solvent Extr. Ion Exch., 2011, 29, 619 CrossRef CAS; (f) D. Kirsanov, A. Legin, M. Tkachenko, I. Surzhina, M. Khaidukova and V. Babain, AIP Conf. Proc., 2011, 1362, 104 CrossRef CAS PubMed.
- M. Y. Alyapyshev, V. A. Babain, Y. A. Pokhitonov and V. M. Esimantovskiy, Adv. Nucl. Fuel Cycles Syst., 2007, 1836 CAS.
- H. Sajiki, T. Ikawa and K. Hirota, Org. Lett., 2004, 6, 4977 CrossRef CAS PubMed.
- (a) T. J. Bartczak, Z. M. Michlska, B. Ostaszewski, P. Sobota and K. Strzelec, Inorg. Chim. Acta, 2001, 319, 229 CrossRef CAS; (b) D. S. Marlin, M. M. Olmstead and P. K. Mascharak, J. Mol. Struct., 2000, 554, 211 CrossRef CAS; (c) A. Itai, Y. Toriumi, S. Saito, H. Kagechika and K. Shudo, J. Am. Chem. Soc., 1992, 114, 10649 CrossRef CAS.
- (a) Q. Y. Yang, Z. Y. Zhou and J. Y. Qi, Acta Crystallogr., 2001, E57, o1161 Search PubMed; (b) M. F. Mayer, S. Nakashima and S. C. Zimmerman, Org. Lett., 2005, 7, 3005 CrossRef CAS PubMed; (c) X. Li, Y. Jia, Y. Ren, Y. Wang, J. Hu, T. Ma, W. Feng and L. Yuan, Org. Biomol. Chem., 2013, 11, 6975 RSC.
- (a) B. Dolenský, R. Konvalinka, M. Jakubek and V. Král, J. Mol. Struct., 2013, 1035, 124 CrossRef PubMed; (b) J. G. Sośnicki and P. E. Hansen, J. Mol. Struct., 2004, 700, 91 CrossRef PubMed.
- Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1996, 118, 7529 CrossRef CAS.
- (a) W. E. Stewart and T. H. Siddall III, Chem. Rev., 1970, 70, 517 CrossRef CAS; (b) N. Ototake, T. Taguchi and O. Kitagawa, Tetrahedron Lett., 2008, 49, 5458 CrossRef CAS PubMed; (c) J. S. Laursen, J. Engel-Andreasen, P. Fristrup, P. Harris and C. A. Olsen, J. Am. Chem. Soc., 2013, 135, 2835 CrossRef CAS PubMed.
- (a) B. Gong, Acc. Chem. Res., 2008, 41, 1376 CrossRef CAS PubMed; (b) L. Yuan, W. Feng, K. Yamato, A. R. Sanford, D. Xu, H. Guo and B. Gong, J. Am. Chem. Soc., 2004, 126, 11120 CrossRef CAS PubMed.
- J. F. Malone, C. M. Murray, G. M. Dolan, R. Docherty and A. J. Lavery, Chem. Mater., 1997, 9, 2983 CrossRef CAS.
- B. Klepetářová, E. Makrlík, V. A. Babain and V. Kašička, Acta Crystallogr., 2012, E68, o1099 Search PubMed.
- (a) N. H. Shah, G. L. Butterfoss, K. Nguyen, B. Yoo, R. Bonneau, D. L. Rabenstein and K. Kirshenbaum, J. Am. Chem. Soc., 2008, 130, 16622 CrossRef CAS PubMed; (b) B. F. Pedersen and B. Pedersen, Tetrahedron Lett., 1965, 34, 2995 CrossRef; (c) R. Yamasaki, A. Tanatani, I. Azumaya, S. Saito, K. Yamaguchi and H. Kagechika, Org. Lett., 2003, 5, 1265 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, N. Rega, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. Martin, L. D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian Inc., Wallingford, CT, 2004.
- (a) M. Ocak, H. Alp, H. Kantekin, H. Karadeniz and Ü. Ocak, J. Inclusion Phenom. Macrocyclic Chem., 2008, 60, 17 CrossRef CAS; (b) H. Alp, M. Ocak, M. Özdemir and Ü. Ocak, Sep. Sci. Technol., 2006, 41, 3039 CrossRef CAS; (c) Ü. Ocak, H. Alp, P. Gökçe and M. Ocak, Sep. Sci. Technol., 2006, 41, 391 CrossRef; (d) Ü. Ocak, Y. Gök and H. B. Şentürk, Sep. Sci. Technol., 2009, 44, 1240 CrossRef.
- P. Kapoor, A. Kumar, J. Nistandra and P. Venugopalan, Transition Met. Chem., 2000, 25, 465 CrossRef CAS.
- M. Iqbal, P. K. Mohapatra, S. A. Ansari, J. Huskens and W. Verboom, Tetrahedron, 2012, 68, 7840 CrossRef CAS PubMed.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995 RSC.
- (a) H. Alp, H. Z. Gök, H. Kantekin and Ü. Ocak, J. Hazard. Mater., 2008, 159, 519 CrossRef CAS PubMed; (b) N. Singh and D. O. Jang, J. Hazard. Mater., 2009, 168, 727 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC.
- Y. Y. Zhu, C. Cui, N. Li, B. W. Wang, Z. M. Wang and S. Gao, Eur. J. Org. Chem., 2013, 17, 3101 CrossRef PubMed.
- J. S. Bae and H. S. Freeman, Fibers Polym., 2002, 3, 140 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization details, extraction data. CCDC 990530 and 990531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra02030h |
| This journal is © The Royal Society of Chemistry 2014 |