
Selective N-alkylation of primary amines with R–NH2·HBr and alkyl bromides using a competitive deprotonation/protonation strategy†
Abstract
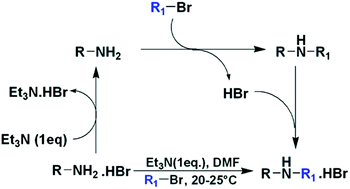
Monoalkylation of primary amines using amine hydrobromides and alkyl bromides has been carried out. Under controlled reaction conditions the reactant primary amine was selectively deprotonated and made available for reaction, while the newly generated secondary amine remained protonated, and did not participate in alkylation further. Reaction was carried out under mild reaction conditions and was applicable to a wide range of primary amines and alkyl bromides.
 Please wait while we load your content...
Please wait while we load your content...

