 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild and rapid surface initiated ring-opening polymerisation of trimethylene carbonate from cellulose†
Samuel A. Pendergraph*,
Gregor Klein,
Mats K. G. Johansson and
Anna Carlmark*
KTH Royal Institute of Technology, School of Chemical Science and Engineering, Fibre and Polymer Technology, Teknikringen 56, SE-100 44, Stockholm, Sweden. E-mail: annac@kth.se; spender@kth.se
First published on 29th April 2014
Abstract
Surface initiated ring-opening polymerisation (SI-ROP) of trimethylene carbonate (1,3-dioxane-2-one, TMC) from cellulose surfaces has been studied for the first time. Specifically, organocatalytic systems employing 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) were implemented in the ROP of TMC, initiated by the hydroxyl groups on the cellulose chain, to form polymer grafts on the surface of filter papers. A sacrificial initiator was added to the reaction solution, resulting in the formation of free, unbound polymer formed in parallel to the surface grafting. The properties of the polymer grafted paper were studied utilising infrared spectroscopy, thermal gravimetric analysis, scanning electron microscopy and contact angle measurements. The free polymers were characterised with nuclear magnetic resonance spectroscopy and size exclusion chromatography. The grafting resulted in hydrophobic papers in as little as one minute and the ability to control the grafting length of the polymer from the surface was demonstrated by either altering the time of the polymerisation or the ratio of free initiator to monomer. This polymerisation route provides milder conditions than conventional metal-catalysed ROP, greatly reduces reaction times and thus is an attractive method for modification of natural biopolymers compared to previously described methods.
Introduction
The grafting of polymers from solid surfaces has been implemented as an effective technique to control the interfacial behaviour.1–7 Different approaches have been examined, which include conventional free radical polymerisations,8–10 as well as controlled techniques such as atom transfer radical polymerisation (ATRP),11–13 nitroxide mediated radical polymerisations (NMP),14,15 reversible addition fragmentation chain transfer (RAFT),16–21 and ring-opening polymerisation (ROP).3,22–28 Controlled surface initiated polymerisation techniques have been applied to create well defined surfaces for a plethora of applications ranging from electronics, reinforced composites, optical devices and drug delivery vehicles.2,4,6,12,23,29,30The modification of cellulosic materials has been a well-studied challenge over the last several decades.23 The high modulus that is inherent for cellulose as well as the natural abundance and availability of the material continues to be the motivation for hybrid cellulosic based materials.23 However, realization of cellulose derived materials in several applications has been hindered due to the poor interfacial compatibility with many synthetic polymers. Conversely, surface polymerisations have been utilised to obviate poor adhesion between these interfaces.1,16,31,32 For example, inclusion of cellulosic materials into fibre reinforced composites has significantly increased in recent years.3,23,24,26,33
The aforementioned controlled polymerisation techniques have been studied in order to obtain versatile and functional surfaces. RAFT and ATRP polymerisations have successfully introduced characteristics on cellulose such as hydrophobicity, responsivity, and formation of precursor reactive groups.1,5,16,17,20,28,32,34,35 However, the utilisation of these synthetic routes requires a modification of the cellulose to introduce the necessary reactive/initiating group. Polyesterification reactions have been widely evaluated on different types of cellulose as they do not require an initial chemical modification of the interface.3,22–28 In this reaction, the native hydroxyl groups in the polysaccharide backbone are utilised as initiators, which is a great advantage with this technique. However, despite several studies on polyester grafting, the reactions are typically focused on metal catalysed polymerisations, for the ROP of lactones and lactides. Hafrén and Cordova utilised an organic acid for the ROP of ε-caprolactone (ε-CL) from a cellulosic substrate; however their procedures required high temperatures.36 Recently, our group has published a report of using a basic organocatalytic system which was employed (1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)), for the grafting of ε-CL from a cellulose model surface.37 The use of these types of catalysts has been demonstrated to create well-controlled solution based polymers with short reaction times and under mild conditions.38
Lactones and lactides have proven to be versatile monomers in ROP from cellulose and demonstrate a potential for biomedical applications.39,40 The advantage of the resulting polyesters is their potential biodegradability as they are able to hydrolyse under in vivo conditions. However, one drawback with polyesters is the generation of acid by the formation of carboxylic acid groups during hydrolysis. Polycarbonates are a class of polymers which demonstrate similar potential for biomedical applications in terms of degradation under in vivo conditions; however they do not generate acidic by-products in the hydrolysis of the polymer.29,41–45 These characteristics have motivated us to study polycarbonates grafted from cellulose interfaces, which to our present knowledge have not been explored previously. In this report, we describe the first polymerisation of trimethylene carbonate (1,3-dioxane-2-one, TMC) from cellulosic fibre surfaces. Our method utilises organocatalytic bases to initiate ROP from paper fibres directly. We demonstrate the ability to create hydrophobic paper with tuneable control of the amount of polymer grafted on the surface. The reactions described herein are performed under milder conditions than previously reported work on ROP of lactones and lactides from cellulosic surfaces. Finally, we demonstrate the ability to create hydrophobic, polycarbonate functionalised papers within a minute at room temperature without prior modification of the cellulosic material.
Experimental
Materials
1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), benzylic alcohol and benzoic acid were purchased from Sigma Aldrich and used as received. Dichloromethane (DCM) was purchased from VWR and used without further purification. Trimethylene carbonate (TMC) was synthesized according to literature procedures.46 Whatman #1 filter paper was first washed with ethanol and DCM thoroughly. Subsequently, the paper was dried in an oven overnight (>12 hours) at 120 °C prior to the grafting polymerisation.Instrumentation
Proton nuclear magnetic resonance (1H-NMR) spectra were recorded using a Bruker AM 400a with deuterated chloroform (CDCl3) as the solvent. Molecular weight (Mn) and dispersity (ĐM) were evaluated with size exclusion chromatography (SEC) with a TOSOH EcoSEC HLC-8320GPC system equipped with an EcoSEC RI detector and three columns (PSS PFG 5 μm, Microguard 100 Å and 300 Å) (molecular weight resolution range: 300–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) from PSS GmbH, using DMF (0.2 mL min−1) containing 0.01 M LiBr as the mobile phase at 50 °C. The measurements were calibrated against a low dispersity poly(methyl methacrylate) (PMMA) standard ranging from 700 to 2000000 g mol−1.
000 g mol−1) from PSS GmbH, using DMF (0.2 mL min−1) containing 0.01 M LiBr as the mobile phase at 50 °C. The measurements were calibrated against a low dispersity poly(methyl methacrylate) (PMMA) standard ranging from 700 to 2000000 g mol−1.
Static contact angles were measured at 50% RH and 23 °C on a KSV instrument CAM 200 equipped with a Basler A602f camera, using a volume of 5 μL of Milli-Q water.
Infrared spectroscopy (IR) was conducted on a Perkin-Elmer Spectrum 2000 FT-IR with a MKII Golden Gate, Single Reflection ATR system from Specac Ltd., London, U.K. All spectra were normalised against a specific ATR crystal absorption (1900–2300 cm−1) in the comparison of the cellulose grafting.
Field emission scanning electron microscopy (FE-SEM) images were recorded on a Hitachi S-4800 FE-SEM and the samples were coated according to previously reported procedures.22
Thermal gravimetric analyses (TGA) were performed on a TGA/DSC 1 Mettler Toledo AG, Analytical Switzerland. Samples were subjected to a heating rate of 10 °C min−1 from 40 to 700 °C under nitrogen atmosphere with a flow rate of 60 mL min−1.
Polymerisation of TMC on cellulose fibres
The conditions for the TMC polymerisation were based on previously reported procedures in the literature.38,47 All polymerisations in the test series followed a general procedure as described below, varying reaction conditions as presented in Table 1. A vial containing a magnetic stir-bar was flame-dried. The papers (2 pieces with dimensions of 1.5 × 0.5 cm) were directly added from the drying oven and the vial was then capped with a rubber septum. The vial was evacuated and backfilled with argon to form a dry, inert atmosphere within the vial. After the vial had cooled down to room temperature, TMC (0.5 g, 4.9 × 10−3 mol) was added to the flask. The flask was subsequently evacuated and backfilled with argon for 10 minutes to remove oxygen and water. A fresh stock solution of benzylic alcohol (8.5 mg mL−1) in DCM was added, under argon, to introduce the sacrificial initiator. For example, in 250:1 reactions, 250 μL of the benzylic stock solution (2.12 mg, 1.96 × 10−5 mol) was added to the mixture. Catalyst stock solution (30 mg mL−1) was added to the reaction mixture and the mixture was allowed to stir for one minute at room temperature to ensure dissolution of the monomer. In all experiments, the monomer to catalyst molar ratio was kept constant at a 100:1 ratio and the sample vials subsequently diluted with dry DCM under argon so the total volume of the reaction mixtures was 850 μL. The reaction mixture was then placed into an oil bath thermostated at 40 °C for a specified time. When the reaction was completed, the solution was quenched with benzoic acid in a 3 molar excess relative to the DBU catalyst. The modified paper was removed from the reaction solution and rinsed with DCM and ultrasonicated in DCM for 5 minutes to remove any physically adsorbed polymer. The solvent from the rinsing was transferred to the remaining reaction mixture and then concentrated in a rotary evaporator. The solution was then poured in an excess of methanol at 0 °C and the precipitated polymer collected for IR, 1H-NMR and SEC analysis. The paper was vacuum dried overnight at 50 °C prior to FTIR, TGA, SEM and contact angle measurements. Experiments using the TBD catalyst were conducted in the same manner as DBU, except that the time was held constant at 10 minutes for all experiments. In a final experiment, the reaction was run for 1 minute and then quenched and processed identically to the other TBD and DBU reactions.
| Sample | Catalyst | Monomer/initiator ratio [M] | Time | Conversion (%) | Mnb (kDa) | Mnc (kDa) | ĐM |
|---|---|---|---|---|---|---|---|
| a No added sacrificial initiator.b Calculated from end-group analysis by 1H-NMR.c Measured by SEC. | |||||||
| 1 | DBU | 250 | 3 h | 86 | 10 | 7.2 | 1.40 |
| 2 | DBU | 250 | 6 h | 92 | 10 | 7.7 | 1.36 |
| 3 | DBU | 250 | 24 h | >99 | 11 | 7.0 | 1.60 |
| 4a | DBU | — | 24 h | >99 | N/A | 61 | 1.43 |
| 5 | TBD | 50 | 10 min | >99 | 5.0 | 4.7 | 1.86 |
| 6 | TBD | 250 | 10 min | >99 | 10 | 6.9 | 1.33 |
| 7 | TBD | 100 | 1 min | >99 | 7.5 | 9.8 | 2.06 |
Results and discussion
The impetus of this study was to evaluate the characteristics of organocatalytic polymerisations of TMC from cellulose fibre surfaces. Filter paper (Whatman #1) has previously proven to be a robust and reliable surface for polymerisations of a plethora of various monomers, owing to its high cellulosic content (>98%) and purity.31 While ROP and ring-opening metathesis polymerisation (ROMP) have been studied from cellulose fibres, limited work has been undertaken on understanding the behaviour of ring-opening reactions, catalysed by metal free organocatalysts.22,37 Furthermore, ROP of cyclic carbonates have not been previously reported from paper based materials. In order to evaluate the polymerisations from cellulose, we employed similar reactions conditions as reported by Waymouth, Hedrick and co-workers (Fig. 1) for polymerisation of TMC in solution, implementing the same catalysts on a heterogeneous system.38,47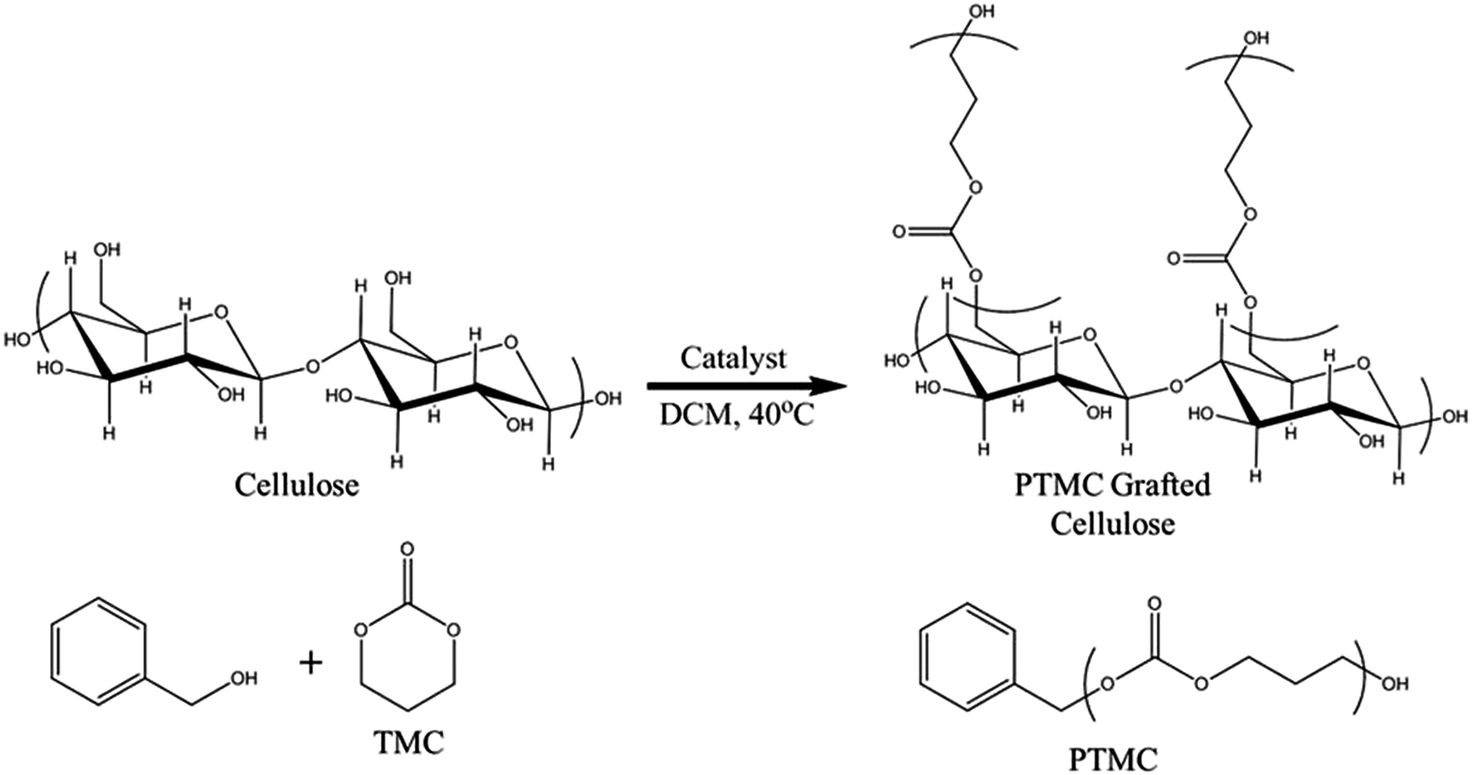 | ||
| Fig. 1 Reaction scheme of the grafting polymerisation from the filter paper. | ||
In most of the grafting reactions, a sacrificial initiator was added to the polymerisation solution, resulting in formation of free, unbound polymers. Previous work on grafting from cellulose utilising ATRP and RAFT have shown that polymer formed in the solution in parallel to the surface initiated ring-opening polymerisation (SI-ROP) has similar properties in terms of Mn and dispersity.16,48 It was anticipated that this would also be valid for SI-ROP of carbonates. Therefore, this methodology was also chosen for these experiments as surface-bound polycarbonates are challenging to cleave off the surface of cellulose fibres by conventional methods (e.g. hydrolysis) since this would also degrade the polymer itself. However, analysing the free polymer formed, and assuming that the free polymer and the surface bound polymer are similar, the molecular weight and dispersity (ĐM) of the brush can be indirectly determined.48
Ring-opening polymerisation with DBU catalyst
The free polymers formed were evaluated by SEC and 1H-NMR analysis, varying the reaction times to obtain the molar masses, conversions, and ĐM for both catalytic systems as presented in Table 1. DBU was utilised to evaluate the molecular weight dependence given a target molecular weight as a function of time. This catalyst was selected because it has previously been established to exhibit slower reaction rates in the ROP of TMC compared to other more active organocatalysts (e.g. TBD).38,47 A relatively slow reaction rate allowed for a more distinct determination of the effect of reaction time (i.e. the reaction kinetics of a very rapid reacting system is difficult to practically assess utilising conventional analytical techniques). The conversion was measured by evaluating the up-field shift in the methylene peaks of the TMC monomer to the polymer, from 4.45 ppm to 4.2 ppm and from 2.2 to 2.0 ppm (an example is provided in Fig. S1†).38,47 Polymerisations catalysed with DBU at room temperature had virtually no conversion after 8 hours under inert conditions. However, when the temperature was raised to 40 °C, the reaction proceeded in a linear manner up to 6 hours (sample 1 and 2) and subsequently neared completion after 6 hours according to the 1H-NMR data. Longer reaction times (sample 3, 24 hours) produced a paper with similar hydrophobic characteristics; however this also lead to a broadening of the ĐM of the free forming polymer, which was consistent with previously reported results.38,47 The ability of cellulose to initiate TMC polymerisation without any sacrificial initiator in the reaction mixture ([TMC]/[DBU] = 100) was also investigated. As no free initiator was added, free polymers were not expected to be formed. However, after precipitation of the reaction solution, a polymer which had higher molecular weight compared to the other reactions could be isolated. We propose that this polymer was formed from remaining water molecules, which could function as initiators, or from other side reactions that could potentially occur, such as chain scission of the propagating brushes, cleaving chains off the surface. It should in this context be noted that it is well known that it is very difficult to completely remove bound water from cellulose due to its hygroscopic structure.48 The procedure used in the present study was, however, in accordance to the state of the art in minimising the water content under conventional laboratory conditions. The high molar mass obtained further implies that a high concentration of monomers compared to free initiator functions was found in this system.The grafted cellulose surfaces were analysed by contact angle measurements, IR spectroscopy, SEM and TGA. Static contact angle measurements were performed on the grafted cellulose and compared to native cellulose surfaces. It should here be noted that the two main factors affecting the macroscopic contact angle determined in this study was the surface energy (surfaces tension) measured on an Ångström-level and the surface topography measured over a length scale ranging from nanometres to micrometers.49 Native cellulose paper had no measureable contact angle due to its highly hydrophilic nature. However, the polycarbonate grafted filter papers exhibited stable static contact angles of 100 ± 3 degrees for reaction times of 3 hours or more for the DBU catalysed reactions as shown in Fig. 2. Prolonged reaction times did not increase the contact angles above these values which imply that this was the intrinsic surface behaviour of PTMC surfaces for this specific surface topology. The contact angles were relatively independent of molecular weight and reaction times indicating that the surface on the Ångström-level was rather homogeneous and above a threshold level above which a chain extension do not contribute to further changes in surface energy. The contact angles of 100° were considerably larger than previously reported literature values for PTMC films (∼75°).41 The discrepancy between our values and thin films of PTMC prepared in literature was due to the roughness in the filter paper. In Fig. 3, SEM images are shown for the unmodified paper and for 24 hours of polymerisation with DBU. The unmodified fibres had a rough exterior due to the fibrillar structure of the cellulose fibres. The polycarbonate grafted filter paper appeared to have a smoother surface and the total paper was less porous overall, which is consistent to our previous observations.22
 | ||
| Fig. 2 Contact angle of a PTMC grafted paper after polymerisation (24 hours) with the DBU catalyst at 250:1 monomer/initiator ratio. | ||
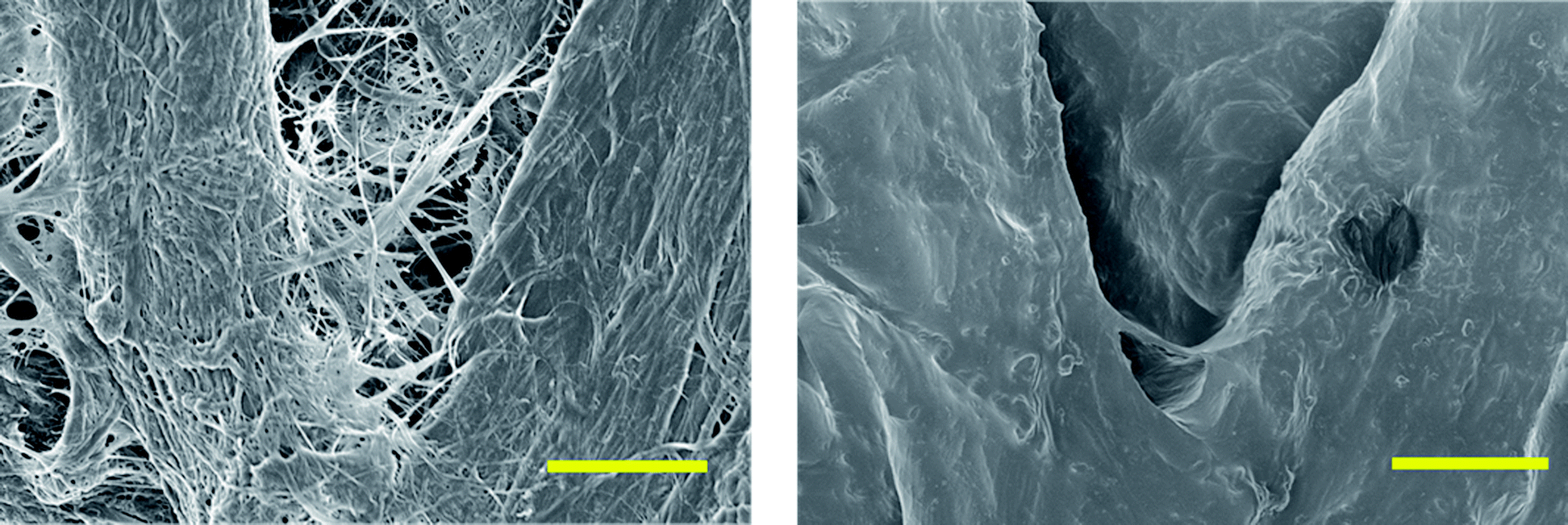 | ||
| Fig. 3 SEM images of unmodified filter paper (left) and PTMC grafted paper (sample 4). The scale bar in both samples is 10 μm. | ||
IR spectroscopy was performed on the grafted filter papers to confirm the attachment of polymers to the surface. Unmodified cellulose does not possess carbonyl functional groups, and therefore they do not have IR adsorption between 1600–1800 cm−1. The reference sample of unmodified filter paper shows no significant peak in this region, as shown in Fig. 4. The pure polycarbonate (PTMC), isolated from the reaction, shows a large stretch vibration around 1750 cm−1, which is characteristic of a carbonate carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration The papers grafted with TMC for 3 hours, exhibited a peak appearing at 1750 cm−1, originating from the carbonate grafted onto the surface. Increasing the polymerisation time to 6 hours, increased the peak in the 1750 cm−1 region, with an even stronger absorption observed for the samples grafted for 24 hours. The increased absorption was indicative of a higher PTMC polymer content on the surface. The increased PTMC content could have arisen from two possibilities. First, the increased time of the reaction allowed for the monomers to propagate, thus creating longer chains assuming a constant grafting density of the chains. Second, continuing reactions of unmodified hydroxyl groups either through grafting-from the surface or PTMC homopolymers grafting-to the surface could have increased the grafting density.38,47 Conversely, we attributed the increase in the carbonyl signal in IR primarily to increasing the molecular weight of the grafted polymer chains due to steric hindrance. Interestingly, this increased amount of polymer on the paper differed from the trend of the data obtained from the SEC and NMR analysis of the free formed polymers, Table 1, entries 1–3. As can be seen, the molecular weights of the free forming polymers are similar in all cases, regardless of the variation in reaction time.
O stretching vibration The papers grafted with TMC for 3 hours, exhibited a peak appearing at 1750 cm−1, originating from the carbonate grafted onto the surface. Increasing the polymerisation time to 6 hours, increased the peak in the 1750 cm−1 region, with an even stronger absorption observed for the samples grafted for 24 hours. The increased absorption was indicative of a higher PTMC polymer content on the surface. The increased PTMC content could have arisen from two possibilities. First, the increased time of the reaction allowed for the monomers to propagate, thus creating longer chains assuming a constant grafting density of the chains. Second, continuing reactions of unmodified hydroxyl groups either through grafting-from the surface or PTMC homopolymers grafting-to the surface could have increased the grafting density.38,47 Conversely, we attributed the increase in the carbonyl signal in IR primarily to increasing the molecular weight of the grafted polymer chains due to steric hindrance. Interestingly, this increased amount of polymer on the paper differed from the trend of the data obtained from the SEC and NMR analysis of the free formed polymers, Table 1, entries 1–3. As can be seen, the molecular weights of the free forming polymers are similar in all cases, regardless of the variation in reaction time.
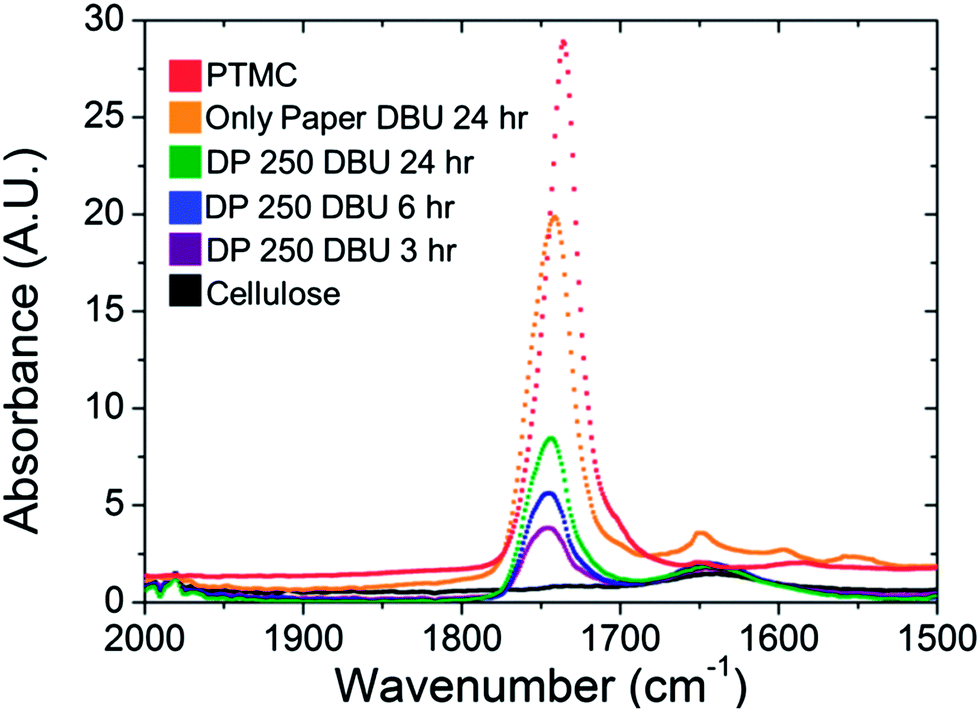 | ||
| Fig. 4 IR spectroscopy on PTMC grafted paper (samples 1–4, Table 1), unmodified cellulose and bulk PTMC (extracted from sample 4). The range displayed illustrates the presence of a carbonyl peak due to the carbonate functionality at 1750 cm−1. | ||
There appeared to be an upper limit to the molecular weight of this polymerisation, which can occur with higher targeted molecular weights of TMC.38,47 It could be that the surface-initiated polymerisation and the free polymerisation do not follow the same reaction conditions, as in the case of SI-ATRP of cellulose, and that side-reactions are occurring at a higher rate in the bulk polymerisation compared to the surface-bound polymerisation.32 In sample 4, no sacrificial initiator was added to the grafting reaction which resulted in a stronger adsorption peak in IR, suggesting that a considerably higher molecular weight polymer had been grafted to the surface in this case. Regardless of the absence of free initiator, a free polymer was formed in bulk, likely initiated from remaining water molecules in the system. As can be seen in Table 1, entry 4, this polymer has considerably higher molecular weight than the ones formed from the free initiator. Furthermore, as this polymer did not contain any benzylic methylene end-groups, no calculations regarding the molecular weight could be performed from 1H-NMR.
The samples polymerised with DBU were analysed with TGA to evaluate the thermal properties of the polycarbonate grafted filter paper in comparison to unmodified papers and PMTC homopolymer. As shown in Fig. 5, there was a significant difference in degradation temperature between the PTMC and the unmodified cellulose paper. PTMC had an onset of thermal degradation temperature of around 240 °C, followed by a precipitous decline in mass. The low temperature for degradation of PTMC has been attributed to the depolymerisation of the carbonate group in the backbone.42 Conversely, unmodified cellulose had the highest degradation temperature, over 100 °C more than PTMC. As expected, when the degree of polymerisation of the grafted polymer increased, the temperature where the onset of thermal degradation occurred was lowered. This was expected; when the mass content of the PTMC was increased, the mass content should begin to show the onset of degradation at a lower temperature. Previous degradation studies with copolymers of PTMC and other polyesters have shown that increasing TMC content shifts the onset of degradation closer towards the pure PTMC degradation temperature.42 Furthermore, despite a significant difference in the degradation temperature of the polyesters and PTMC, they observed a singular decrease in the mass and not a multi-step system.42
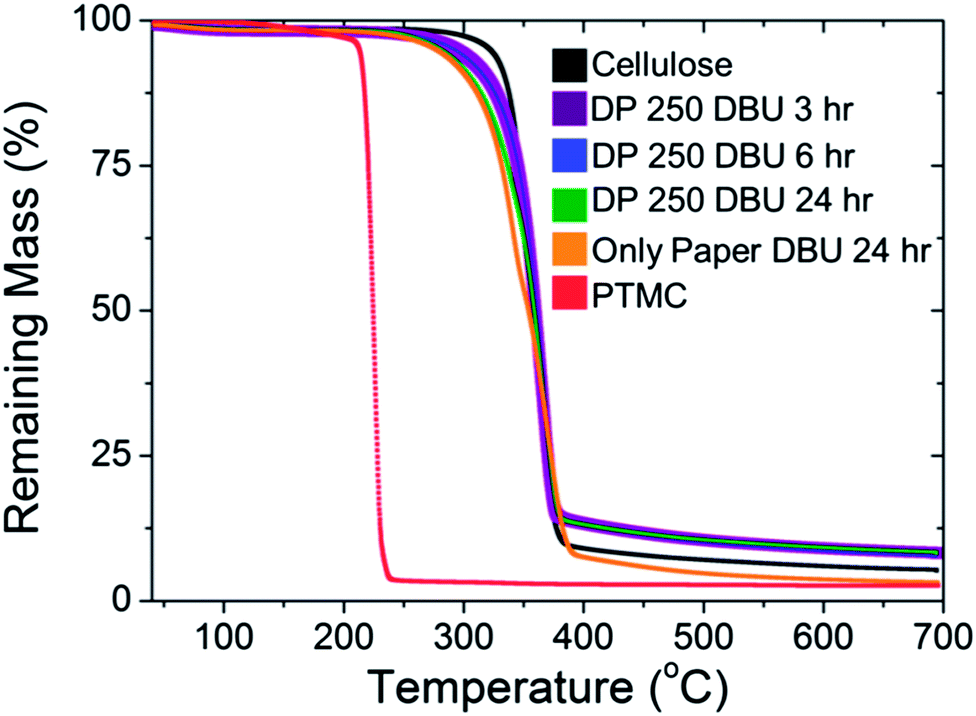 | ||
| Fig. 5 TGA of unmodified cellulose, bulk PTMC (from sample 4) and PTMC grafted paper (samples 1–4, Table 1). | ||
Rapid polymerisation of cellulose utilising TBD
The polymerisation of TMC from cellulose was controllable with the use of DBU as a catalyst over several hours, however the polymerisation times required were around 8 hours for total conversion of the monomer. In order to reduce the reaction times, a more active catalyst, TBD, was also investigated for the SI-ROP of TMC.38,47 In the experiments, the polymerisation with TBD had conversions of over 99% in less than 10 minutes. Therefore, another strategy to control the molecular weight grafted to the paper was implemented. Using TBD as the catalyst, the target molecular weight was adjusted rather than the reaction time. Three different molecular weights were targeted by altering the amount of sacrificial initiator.As shown with entries 5 and 6 in Table 1, the resulting free forming polymer characteristics are shown. The increase in dispersity for the DP of 50 versus 250 was attributed to the rapid conversion and subsequent chain transfer reactions which broaden the ĐM, similar to sample 3 in the DBU polymerisation.38,47 The polycarbonate grafted papers utilising TBD as catalyst were analysed in the same manner as the ones catalysed by DBU. In Fig. 6, the IR spectra of all the grafted filter papers are shown and compared to neat filter paper and PTMC. The carbonate peak at 1750 cm−1 was increasing with higher target molecular weight after 10 minutes of polymerisation. The reduction in the sacrificial initiator produced higher molecular masses on the paper. The filter paper possessed comparable hydrophobic contact angle characteristics to the DBU catalysed reactions.
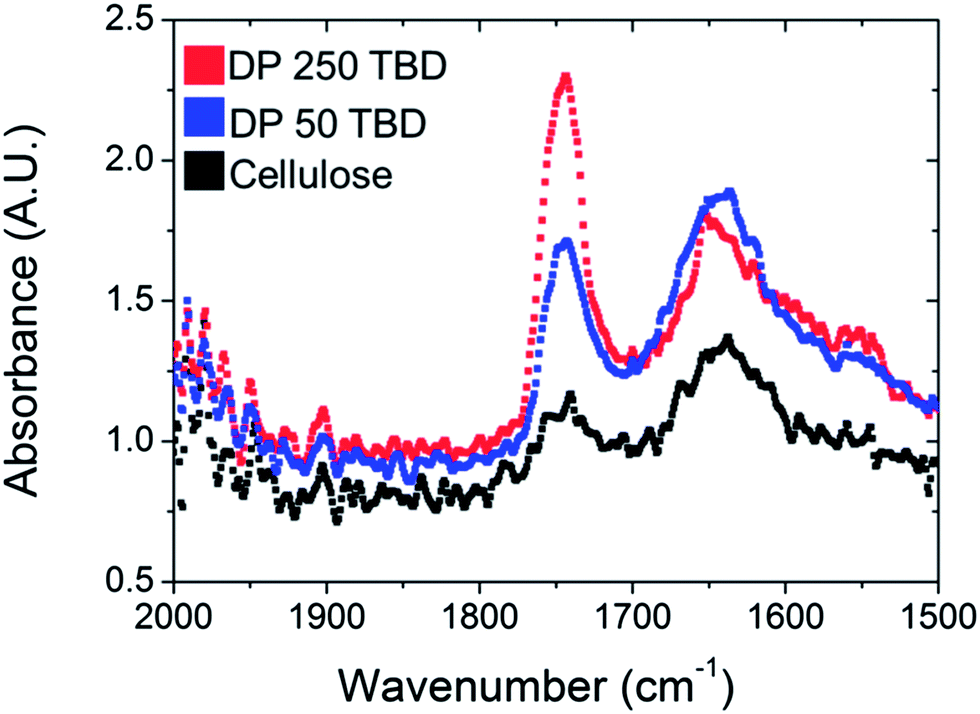 | ||
| Fig. 6 IR spectroscopy of PTMC grafted papers (samples 5 and 6, Table 1) and unmodified paper. | ||
However, we wanted to explore how quickly the polymerisation could be implemented for the paper to become hydrophobic. Another reaction was run for 1 minute (target DP = 100) with TBD and subsequently quenched. The reaction was run to over 99% conversion confirmed by NMR. The grafting of the paper was confirmed by IR spectroscopy (Fig. S2†), where there is a sharp increase in absorption at 1750 cm−1. After processing the paper, the sample was tested for contact angle; the paper was equally as hydrophobic (104 ± 3) as the previous DBU samples after 24 hours (Fig. 7). The ability for unmodified paper to become transformed in very little time can potentially enable the use of functional polycarbonates to create new applications from cellulosic materials. We anticipate these findings to be a valuable procedure to not only introduce new functional groups, but to also quickly introduce interesting polymer architectures off of cellulosic interfaces.
 | ||
| Fig. 7 Contact angle of PTMC grafted paper after 1 minute of reaction time and subsequent quenching of the polymerisation. | ||
Conclusions
Herein, the first SI-ROP of carbonates from cellulose surfaces was reported. Organocatalysts were implemented to demonstrate controlled polymerisation with tuneable grafting lengths on the paper. The polymerisations can be implemented without prior functionalization of the cellulose. Ring-opening polymerisations of TMC resulted in hydrophobic papers and the grafted polymer on the surface could be tailored through two different routes. The grafting content could be tuned by adjusting the reaction time when DBU catalyst was employed or by varying the ratio of monomer to initiator when utilising the more active TBD catalyst. Rapid grafting polymerisations (1 min) were achieved with the use of TBD on filter paper. Implementing organocatalysts with TMC allows a biomedical relevant polymer to be grafted to the surface of a cellulose substrate without metal impurities in the system. The use of cyclic carbonate monomers with TBD can be used as a potential strategy to easily and rapidly introduce functionality onto unmodified cellulosic substrates under mild conditions. The combination of the speed of the reaction and the preclusion modification steps in paper prior to polymerisation can enable this as a potential synthetic route for industrial applications.Acknowledgements
The authors greatly acknowledge the KAMI foundation for financial support.Notes and references
- A. Aied, Y. Zheng, A. Pandit and W. X. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 826–831 CAS.
- C. J. Fristrup, K. Jankova and S. Hvilsted, Soft Matter, 2009, 5, 4623–4634 RSC.
- H. Lönnberg, K. Larsson, T. Lindström, A. Hult and E. Malmström, ACS Appl. Mater. Interfaces, 2011, 3, 1426–1433 Search PubMed.
- S. H. Park, H. S. Lee, J. D. Kim, D. W. Breiby, E. Kim, Y. D. Park, D. Y. Ryu, D. R. Lee and J. H. Cho, J. Mater. Chem., 2011, 21, 15580–15586 RSC.
- J. O. Zoppe, Y. Habibi, O. J. Rojas, R. A. Venditti, L. S. Johansson, K. Efimenko, M. Österberg and J. Laine, Biomacromolecules, 2010, 11, 2683–2691 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS.
- R. C. Advincula, J. Dispersion Sci. Technol., 2003, 24, 343–361 CrossRef CAS PubMed.
- B. de Boer, H. K. Simon, M. P. L. Werts, E. W. van der Vegte and G. Hadziioannou, Macromolecules, 2000, 33, 349–356 CrossRef CAS.
- F. Khelifa, S. Ershov, Y. Habibi, R. Snyders and P. Dubois, ACS Appl. Mater. Interfaces, 2013, 5, 11569–11577 CAS.
- D. Plackett, K. Jankova, H. Egsgaard and H. S. Hvilsted, Biomacromolecules, 2005, 6, 2474–2484 CrossRef CAS PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- J. Pyun, T. Kowalewski and K. Matyjaszewski, Macromol. Rapid Commun., 2003, 24, 1043–1059 CrossRef CAS.
- C. Bartholome, E. Beyou, E. Bourgeat-Lami, P. Chaumont, F. Lefebvre and N. Zydowicz, Macromolecules, 2005, 38, 1099–1106 CrossRef CAS.
- C. Bartholome, E. Beyou, E. Bourgeat-Lami, P. Chaumont and N. Zydowicz, Macromolecules, 2003, 36, 7946–7952 CrossRef CAS.
- M. Barsbay, G. Guven, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and L. Barner, Macromolecules, 2007, 40, 7140–7147 CrossRef CAS.
- C. Li, J. Han, C. Y. Ryu and B. C. Benicewicz, Macromolecules, 2006, 39, 3175–3183 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- D. Roy, J. T. Guthrie and S. Perrier, Macromolecules, 2005, 38, 10363–10372 CrossRef CAS.
- D. Roy, J. S. Knapp, J. T. Guthrie and S. Perrier, Biomacromolecules, 2008, 9, 91–99 CrossRef CAS PubMed.
- M. Semsarilar, V. Ladmiral and S. J. Perrier, J. Polym. Sci., Part A: Polym. Chem, 2010, 48, 4361–4365 CrossRef CAS.
- L. Carlsson, E. Malmström and A. Carlmark, Polym. Chem., 2012, 3, 727–733 RSC.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS PubMed.
- Y. Habibi, A. L. Goffin, N. Schiltz, N. E. Duquesne, P. Dubois and A. Dufresne, J. Mater. Chem., 2008, 18, 5002–5010 RSC.
- M. Labet and W. Thielemans, Cellulose, 2011, 18, 607–617 CrossRef CAS.
- M. Labet and W. Thielemans, Polym. Chem., 2012, 3, 679–684 RSC.
- H. Lönnberg, Q. Zhou, H. Brumer, T. T. Teeri, E. Malmström and A. Hult, Biomacromolecules, 2006, 7, 2178–2185 CrossRef PubMed.
- E. Östmark, D. Nyström and E. Malmström, Macromolecules, 2008, 41, 4405–4415 CrossRef.
- A. C. Engler, J. M. W. Chan, K. Fukushima, D. J. Coady, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2013, 2, 332–336 CrossRef CAS.
- S. C. M. Fernandes, P. Sadocco, A. Aonso-Varona, T. Palomares, A. Eceiza, A. J. D. Silvestre, I. Mondragon and C. S. R. Freire, ACS Appl. Mater. Interfaces, 2013, 5, 3290–3297 CAS.
- A. Carlmark and E. Malmström, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef CAS PubMed.
- S. Hansson, T. Tischer, A. S. Goldmann, A. Carlmark, C. Barner-Kowollik and E. Malmström, Polym. Chem., 2012, 3, 307–309 RSC.
- E. Espino-Perez, J. Bras, V. Ducruet, A. Guinault, A. Dufresne and S. Domenek, Eur. Polym. J., 2013, 49, 3144–3154 CrossRef CAS PubMed.
- A. Carlmark and E. E. Malmström, Biomacromolecules, 2003, 4, 1740–1745 CrossRef CAS PubMed.
- T. Tischer, A. S. Goldmann, A. K. Linkert, V. Trouillet, H. G. Borner and C. Barner-Kowollik, Adv. Funct. Mater., 2012, 22, 3853–3864 CrossRef CAS.
- J. Hafrén and A. Cordova, Macromol. Rapid Commun., 2005, 26, 82–86 CrossRef.
- L. Carlsson, S. Utsel, L. Wågberg, E. Malmström and A. Carlmark, Soft Matter, 2012, 8, 512–517 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- A. Lendlein and R. Langer, Science, 2002, 296, 1673–1676 CrossRef PubMed.
- O. S. Kluin, H. C. van der Mei, H. J. Busscher and D. A. Neut, Biomaterials, 2009, 30, 4738–4742 CrossRef CAS PubMed.
- Y. Marquez, L. Franco and J. Puiggali, Thermochim. Acta, 2012, 550, 65–75 CrossRef CAS PubMed.
- J. Mindemark, J. Hilborn and T. Bowden, Macromolecules, 2007, 40, 3515–3517 CrossRef CAS.
- H. Wang, J. H. Dong, J. K. Y. Qiu and Z. W. Gu, J. Polym. Sci., Part A: Polym. Chem, 1998, 36, 1301–1307 CrossRef CAS.
- K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Macromolecules, 1991, 24, 1736–1740 CrossRef CAS.
- T. Ariga, T. Takata and T. Endo, J. Polym. Sci., Part A: Polym. Chem, 1993, 31, 581–584 CrossRef CAS.
- F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 153–160 CrossRef CAS PubMed.
- S. Hansson, V. Trouillet, T. Tischer, A. S. Goldmann, A. Carlmark, C. Barner-Kowollik and E. Malmström, Biomacromolecules, 2013, 14, 64–74 CrossRef CAS PubMed.
- L. C. Gao and T. J. McCarthy, Langmuir, 2009, 25, 14105–14115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: An example 1H-NMR of the PTMC polymerisation and IR spectroscopy of the paper after 1 minute polymerisation are shown in the ESI. See DOI: 10.1039/c4ra01788a |
| This journal is © The Royal Society of Chemistry 2014 |