
Preparation of an ABC tricyclic model of the cylindrospermopsin alkaloids via a biomimetically inspired pathway†
Abstract
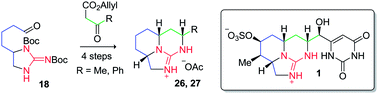
Two tricyclic guanidine model compounds 28 and 29 have been prepared in 12 steps from 1,5-pentanediol using a biomimetic synthetic approach. The pivotal reaction in the sequence is a tethered Biginelli condensation between 21/22 and β-keto esters 23 and 24.
 Please wait while we load your content...
Please wait while we load your content...

