Statistical single-cell analysis of cell cycle-dependent quantum dot cytotoxicity and cellular uptake using a microfluidic system†
Jing Wuab,
Haifang Li*a,
Qiushui Chena,
Xuexia Lina,
Wu Liua and
Jin-Ming Lin*a
aDepartment of Chemistry, Beijing Key Laboratory of Microanalytical Methods and Instrumentation, Tsinghua University, Beijing 100084, China. E-mail: jmlin@mail.tsinghua.edu.cn; lihaifang@mail.tsinghua.edu.cn; Fax: +86 10 62792343; Tel: +86 10 62792343
bSchool of Science, China University of Geosciences (Beijing), Beijing 100083, China
First published on 24th April 2014
Abstract
This paper reports a novel method for the statistical analysis of quantum dot (QD) cytotoxicity and cellular uptake based on single cell cycles, which is part of a series of works on the study of QD cytotoxicity using a microfluidic system (Lab Chip, 2012, 12, 3474–3480; 2013, 13, 1948–1954). The specially designed microfluidic system consisted of a polydimethylsiloxane (PDMS) microwell array for single-cell arrangement and microchannels for QD solution diffusion, enabling effective control of stable cell density and the interdistance between them, as well as maintaining a constant QD concentration with no disturbance of the fluids which can affect cellular uptake. We showed that the treatment of QDs had no influence on cell cycles. However, the QD cytotoxicity was found to be dependent on cellular uptake in various cell cycle phases, because the accumulation and dilution of QDs happened in single cell cycles. The rank of QD cytotoxicity was G2/M > S > G0/G1. Thus, this technology could serve as a new strategy to investigate otherwise inaccessible mechanisms governing nanoparticle cytotoxicity.
Introduction
Nanomedicine is increasingly emerging as a tremendously promising strategy for the therapy and imaging of major disease using nanoparticles (NPs). Quantum dots (QDs) are of great interest and have been extensively utilized in cellular imaging and drug delivery due to their outstanding fluorescence properties1–3 and small size.4–6 However, the cytotoxicity of QDs has become a major obstacle in the universal application of nanomedicine, thus it has attracted significant attention in recent years.7–10Recently, cellular uptake of NPs was reported to be influenced by the cell cycle11 and cell-to-cell interactions.12 The cellular uptake of NPs also depended on the properties of cellular protein corona–NP complexes.13–15 On the other hand, the cellular microenvironment, such as the cell-to-cell distance and NP fluid conditions, might also affect the cellular uptake. Typically, the cytotoxicity of QDs is commonly studied using an average result from cell populations,16–18 thus it is challenging to deeply understand the complex mechanisms of QD cytotoxicity. For these reasons, the statistical analysis of cellular uptake based on single cell cycles will be particularly important in the investigation of QD cytotoxicity under a precisely defined microenvironment. Recently, integrated microfluidic technology has been considered as a powerful tool for spatially and temporally controlling cell growth and stimuli, as well as providing unique advantages such as long term cell culture and high-throughput analysis.19–22 For example, a series of cell densities could be generated23 and cells could be precisely paired to study cell fusion on the microfluidic chip.24 A microfabricated device also worked as a platform to provide a novel way for probing single-cell behavior without flow-induced shear stress.25,26 To our knowledge, the statistical assays of QD cytotoxicity by considering single cell cycles on a well-defined platform will show great significance.
We have developed a microfluidic cell-culture system to exploit the statistical analysis of QD cytotoxicity and cellular uptake based on single cell cycles. The design of a polydimethylsiloxane (PDMS) microwell array for single-cell arrangements allowed effective control of stable cell density and how close the cells were, which was critical for the statistical analysis of cellular uptake. Besides that, a microfluidic structure was specially designed for the diffusion of the QD solution from the side channels to the central one, enabling a constant QD concentration to be maintained and to avoid the influence of the fluids on the cellular uptake. This novel technique for well-defined cellular microenvironments was considered to be a powerful strategy to construct a stable and controllable platform for QD cytotoxicity assays based on single cell cycles.
Experimental
Fabrication of microfluidic device
Standard soft lithography and replica molding techniques were used to fabricate the PDMS microwell array which was utilized for single cell capture. Briefly, a silicon wafer (Tianjin, China) was cleaned using a piranha solution before a negative photoresist SU-8 2050 (Microchem, Newton, MA) was spun onto it at a speed of 3000 rpm to assist the micropillar attachment onto the silica wafer. After baking until it was dry, the photoresist coating was put under UV light exposure without a mask. Then, negative photoresist SU-8 2015 was spin-coated onto it at the same speed to control the depth of microwell to be ∼20 μm. Re-exposure technology was used to fabricate different heights of the microchannel on the upper PDMS. Negative photoresist SU-8 2007 was used to generate the layer of low microchannels and SU-8 2050 was used to manufacture the layer of high main channels. Both of the two molds were finished using the following steps: UV light exposure, development and silanization. The PDMS prepolymer and curing agent (Dow Corning, Sylgard 184, Midland, MI, USA) were premixed 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (by mass) and poured onto the molds. The mixture was degassed under vacuum for 0.5 h and put into an oven for curing at 75 °C for 2 h. The PDMS was peeled off carefully and cut into the designed shape. The inlets and outlets of the microchannels were made using a flat-tipped syringe needle. The two PDMS replicas were sealed together via oxygen plasma (PDC-32 g, Harrick Plasma, Ithaca, NY, USA) treatment for 90 s. The device was sterilized under UV light for 5 min before use.
:1 (by mass) and poured onto the molds. The mixture was degassed under vacuum for 0.5 h and put into an oven for curing at 75 °C for 2 h. The PDMS was peeled off carefully and cut into the designed shape. The inlets and outlets of the microchannels were made using a flat-tipped syringe needle. The two PDMS replicas were sealed together via oxygen plasma (PDC-32 g, Harrick Plasma, Ithaca, NY, USA) treatment for 90 s. The device was sterilized under UV light for 5 min before use.
Operation of single cell array on the microfluidic device
Confluent HepG2 cells in a 60 mm diameter Petri dish (Cancer Institute & Hospital Chinese Academy of Medical Science, Beijing, China) were trypsinized and the cell suspension was collected. The suspension was centrifuged and the supernatant was removed. The remaining cells were resuspended in 100 μL cell culture medium and infused into channel 2 which covered the PDMS microwell array. The microfluidic device had been kept still for 3 min to make the cells fall into the microwells and surplus cells were flushed out using the cell culture medium. Single cell capture was observed and analyzed using a Leica DMI 4000 B fluorescence microscope (Wetzlar, Germany).The captured cells continued to be cultured on the microfluidic device in an incubator for 1–3 days. The cell viability was detected using a live/dead assay kit (calcein-AM/EthD-1, Invitrogen, CA, USA) to show that there was no perturbation of the microfluidic device. Fluorescence images were taken using the same microscope and the data were analyzed using the program Image-Pro Plus 6.0.
Cell cycle assay on flow cytometry
For the cell cycle assay, 60 mm diameter plates of confluent HepG2 cells were divided evenly into four 35 mm diameter plates and then cultured to an abundance of 80%. The QD solution (5 μg mL−1) was added into two Petri dishes and the other two Petri dishes without any treatment were used for control measurements. After 24 h, all of the HepG2 cells were harvested using 0.05% trypsin–ethylenediaminetetraacetic acid and the cell cycles were studied using total DNA staining for flow cytometry. The cells were fixed with 70% ice-cold ethanol for 1 h at 4 °C and centrifuged. The collected cells were rinsed with phosphate buffered saline (PBS) and resuspended in 500 μL PBS containing 50 μg mL−1 propidium iodide (PI), 50 μg mL−1 RNase A and 3.8 mM sodium citrate for incubation (30 min).Cell cycle and QD cytotoxicity
After being cultured on the microfluidic device for 1 day, the captured cells were treated with QDs for 24 h under static and dynamic conditions. A stock solution of QDs (5 mg mL−1) was serially diluted with the cell culture medium to obtain the desired concentration (5 μg mL−1) and was infused into channel 1. As a blank solution, the cell culture medium was infused into channel 3. The two kinds of solutions were diffused into channel 2 through the low microchannels under static conditions. For the dynamic condition, the QD solution (5 μg mL−1) and cell culture medium were injected into channel 1 and 3 respectively at a speed of 0.6 μL min−1 using a syringe pump (Harvard Apparatus PHD 2000, Holliston, MA). Intracellular reactive oxygen species (ROS) and glutathione (GSH) were detected as two indexes of QD cytotoxicity. 10 μM Hoechst 33342 (Invitrogen, CA, USA) and 100 μM dihydroethidium (DHE) (Beijing, China) were used to stain the cells simultaneously for ROS analysis in different cell cycle phases. Fluorescence images were taken twice at the same position under two different exciting wavelengths (λex = 340–380 nm and λex = 535 nm) with a fluorescence microscope equipped with a cooled CCD camera with software of Leica Application Suite, LAS V2.7. Intracellular GSH variation of the cells in different cycle phases was also detected using a similar method but with cells that were stained with Hoechst 33342 and 2,3-naphthalenedicarboxaldehyde (NDA) (Tokyo, Japan) at the same time. The cell cycle was analyzed by Origin software, and the program QCapture Pro (Version 5.1.1.14, Media Cybernetics, USA) was used to analyze the fluorescence intensity of the intracellular ROS and GSH.Results and discussion
Design of the microfluidic device
The microfluidic device was fabricated using re-exposure technology and multi-layer soft lithography. The designed microdevice consisted of two functional components (Fig. 1A): (1) a PDMS microwell array substrate for capturing single cells with controllable cell density and interdistance between the cells, and (2) overlaid PDMS microchannels with different heights for maintaining stable QD concentration and culturing single cells without shear stress. The diameter and depth of the microwells were designed to be 25 μm and 20 μm (Fig. 1B), respectively, which were suitable to capture the HepG2 cells. The microchannels on the top layer included two parts: three main channels and the connecting microchannels. The QD solution and cell culture medium could diffuse from channel 1 and 3 into channel 2 through the lower connecting microchannels (Fig. 1C). There are two different lengths (1.5 mm and 5.0 mm) of the low microchannels to illuminate the impacts of diffusion distance and concentration-dependence of the QD cytotoxicity. The solution concentration in channel 2 was kept constant without any dilution by the two side solutions after a few hours of diffusion, i.e. the cells in microwells were cultured under dilute-free conditions. It was very critical to eliminate the effect of fluids on the cellular uptake and generated shear stress.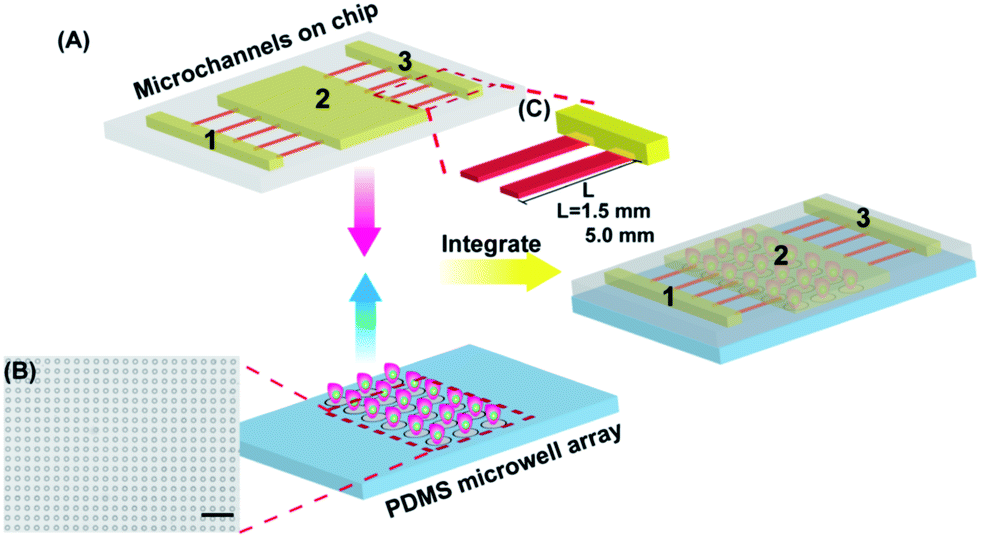 | ||
| Fig. 1 Schematic illustration of the microfluidic device for the statistical analysis of QD cytotoxicity based on single cell cycle observation. (A) The two functional components: PDMS microwell array and microchannels on the chip. The microfluidic device is a combination of both of them. (B) The microscope image of the PDMS microwell array (scale bar: 250 μm). (C) Schematic representation of the microchannels with different heights on the chip. The length of lower microchannels was designed to be 1.5 mm and 5.0 mm. | ||
Single cell array on the microfluidic device
A single-cell array is particularly useful to study cellular heterogeneity.27,28 The depth of the microwells was controlled to be 20 μm which was comparable to the diameter of the HepG2 cells (∼16 μm). More than one cell would be contained in the deeper microwells while cells in the shallow ones could not be confined in them during the operation process. The interdistance between the cells could be regulated through controlling the interwell distance, which was 50 μm in this case. During loading, the cells were filled into channel 2 and passed through the microwell array (Fig. 2A). Actually, different microwell diameters of 25, 30, 35 and 40 μm were tested in this system. For the microwell with a diameter of 25 μm, the overall cell occupancy was approximately 90% and almost all of the occupied microwells included only one cell (Fig. 2B and C). The microwells with diameters of 30 and 35 μm have higher overall cell occupancy while the number of microwells with two to four cells also increased significantly. The cells conveniently entered into larger microwells but they were also easily flushed out, so the number of null microwells led to a low cell occupancy for the microwell with a diameter of 40 μm (Fig. 2C and S1†). As a result, a microwell size of 25/20 μm (diameter/depth) was finally selected to conduct the statistical analysis of QD cytotoxicity. The microwell array was 20 × 150 and the volume of channel 2 was 6 μL. The loading efficiency was about 80%, making sure that the cell concentration in channel 2 reached as high as ∼108 mL−1 and was satisfied throughout this study. The concentration of single cells could be regulated through designing the number of microwells and the size of channel 2. | ||
| Fig. 2 Single cell array on the microfluidic device. (A) Schematic representation of the operation process for generating a single cell array. (B) Fluorescence image of the single cell array (scale bar: 200 μm). (C) Cell distribution with different sizes of PDMS microwells. (D) Cell viability detection on the microfluidic device during 1–3 days. | ||
The captured single cells were further cultured in the microwells. The cell viability was detected using a live/dead (calcein AM/EthD-1) assay kit. The results indicated that the viability of the HepG2 cells was kept above 90% during 1–3 days (Fig. 2D) and showed that the operation of the generated single cell array and the microwells had no adverse impact on the cell viability. A non-perturbing single cell array generation mechanism was provided and the interdistance and concentration of the single cells were controllable on this microfluidic device.
Simulation of QD concentration in the microchannels
To protect the cells from shear stress, the solutions were controlled to diffuse from the side channels into the middle channel which covered the single cell array. The software COMSOL Multiphysics was used to simulate the process under static and dynamic conditions (Fig. 3A and S2†). The low microchannels were designed to be 1.5 mm and 5.0 mm long to investigate how the diffusion distance affected QD cytotoxicity. A balanced concentration in channel 2 was reached after both of the microfluidic devices had been under static conditions for a few hours. It is worthwhile to point out that the time taken to reach a balanced concentration was different: 5 h for the 1.5 mm microfluidic device while it took as long as 10 h for the 5.0 mm microfluidic device. Compared to the long microchannels, the concentration is higher in the short microchannels at the same time point. Data in ESI Fig. S2† also show this tendency. Under dynamic conditions, the concentration in channel 2 had no obvious variation throughout 24 h which is higher than that under static conditions. Amaranth and fluorescein sodium (100 μM) solutions were used to observe the diffusion process. The microscope images were in accordance with the simulated results (Fig. 3B and C). The results indicate that the concentration of the solutions in channel 2 could remain invariable under both static and dynamic conditions. All of the single cells were cultured in a fluid disturbance- and shear stress-free microenvironment.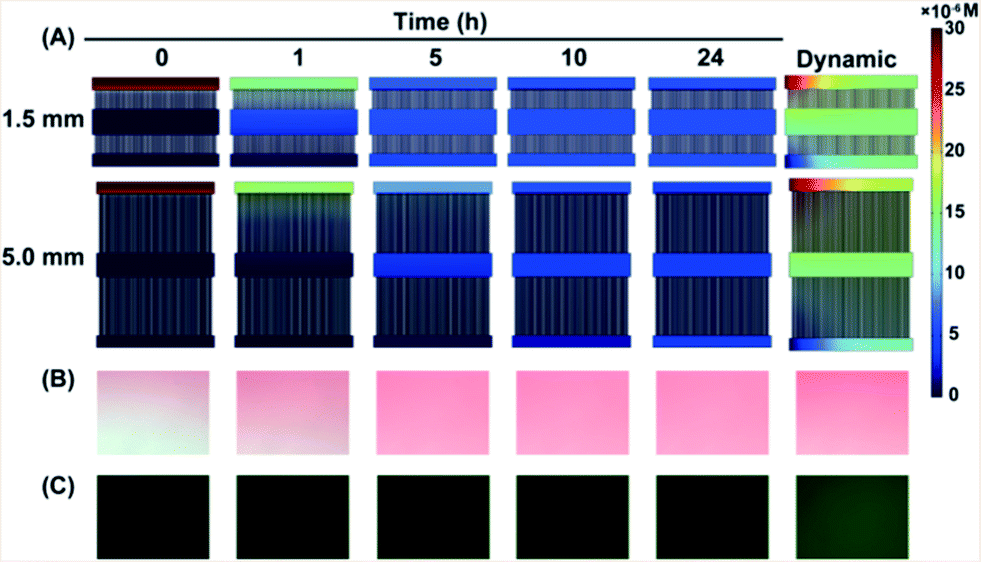 | ||
| Fig. 3 The concentration of the solutions in the microchannels remains constant under static and dynamic conditions. (A) Concentration simulated using Software COMSOL Multiphysics. Amaranth (B) and fluorescein sodium (C) solutions diffused in channel 2, observed using a microscope. | ||
The effect of QD cytotoxicity on cell cycle
In order to analyze the impact of QDs on cell cycle distribution, DNA staining with PI was analyzed using a flow cytometer. The fluorescence intensity of PI was proportional to the quantity of intracellular DNA, according to the cell cycle which was divided into the G0/G1, S and G2/M phases. For cells treated with QDs, the population distributed in G0/G1, S and G2/M phases is 64.21%, 22.80% and 12.99%, respectively. The population of the cells without any treatment distributed in G0/G1, S and G2/M phases is 67.54%, 21.09% and 11.38% (Fig. 4 and Table 1). There is no obvious change in the population of the cells treated with QDs in different phases compared to the control cells. The type and concentration of QDs used here do not perturb the cell cycle distribution and are suitable for studying the effect of cell cycle on QD cytotoxicity. | ||
| Fig. 4 Cell cycle of QD treated and control cells analysis using flow cytometery. | ||
| Cell cycle | QD treated cells | Control cells |
|---|---|---|
| G0/G1 | 64.21% | 67.54% |
| S | 22.80% | 21.09% |
| G2/M | 12.99% | 11.38% |
Role of cell cycle on QD cytotoxicity
The relationship between cell cycle and QD cytotoxicity was investigated and an assay was carried out on the constructed microfluidic platform. ROS (generation) and GSH (reduction) as cellular signal molecules representing a redox state were detected as two cytotoxic indexes in this work.29–31 DHE and NDA were used as two specific fluorescent probes to detect them. Generally, the quantity of intracellular ROS generation remained at a low level and could be easily neutralized by GSH and other antioxidant enzymes. However, QDs are outstanding at transferring energy and aid ROS generation and GSH reduction so the fluorescence of the ROS was enhanced while the fluorescence of GSH was weakened in the presence of QDs.The cell cycle is considered to be an important cellular factor in the biological process. The cell cycle could be divided into four different phases: G1, S, G2 and M. The main task in the G1 phase for the cells is to synthesize RNA and ribosome, and the cell volume is remarkably increased. Cells in the G1 phase prepare levels of nutrition and energy to enter into the S phase. DNA is synthesized in the S phase and protein is fabricated in the G2 phase. After the three phases, the cells enter the M phase and start to divide. The two daughter cells restart a new cell cycle by commencing with the G1 phase. To identify the cell cycle, the cells were simultaneously stained with Hoechst 33342 and DHE or NDA and fluorescence images were taken under the same conditions using fluorescence microscopy. Hoechst 33342 is a fluorescent probe that is specific to nucleic acid staining so the integrated intensity of the same area on the fluorescence images was proportional to the quantity of nucleic acid relative to the cell cycle phases. The nucleic acid of the cells in different phases varies in quantity: G0/G1 phase is 2N, G2/M phase is 4N and S phase falls in between. It was reasonable to infer that the highest integrated intensity should be assigned to the cell in the G2/M phase while the lowest should be assigned to the cell in the G0/G1 phase and the integrated intensity of the cell in the S phase should be in the middle. As a result, the two specific fluorescent probes which simultaneously stained the cells could identify the degree of cytotoxicity of the single cells in the different cell cycle phases and recognize the relationship between cell cycle and QD cytotoxicity.
The fluorescence images in Fig. 5B show that the fluorescence of nucleic acid went in the order G2/M > S > G0/G1 and the fluorescence of the ROS varied with the same sequence. Any redundant ROS reacted with intracellular GSH so the fluorescence intensity of GSH of the cells in different cell cycle phases is arranged in the opposite order (Fig. 5B and 6). As shown in Fig. 5C and D, the curves of the fluorescence intensity variation of cellular ROS in different cell cycle phases exhibit the above tendency for both of the two kinds of microfluidic device. Varying cellular uptake of QDs in different cell cycle phases11 is explained as the main reason for the differentiation of QD cytotoxicity. The rise of membrane tension of metaphase cells leads to the dramatic inhibition of endocytosis. The morphology of the cells also changes during the cell cycles, while the total membrane area and surface expression of membrane-bound proteins vary during cell division.32 Possibly, the cell cycle influences cellular uptake for a series of events which happen during the four phases.32–34 QDs are internalized through autophagy and remain in lysosomes.13 The uptake rates in different cell cycle phases are similar and cell export is negligible once the QDs were internalized.11 However, the uptake time for cells in different cell cycle phases is not the same. The cells in the G2/M phase had the longest uptake time and for that reason they did not divide and the intracellular QDs were not diluted into the cells of the next generation. The cells in the G0/G1 phase had the shortest uptake time, they finished division and almost had no time to accumulate more QDs. The uptake time of the cells in the S phase ranks between the above two phases and the cells internalized some QDs after cell division. As a result, the QD accumulation in the cells of the different phases ranks in the sequence G2/M > S > G0/G1 (Fig. 5A) and the different intracellular QD concentrations lead to various cytotoxic effects. The two different lengths of low microchannels (1.5 mm and 5.0 mm) were designed and the rule was reproved for both of the microfluidic devices (Fig. 5C and D and 6).
 | ||
| Fig. 5 The response of single cells in different cell cycle phases to QD cytotoxicity. (A) Schematic representation of QD uptake of cells in different cell cycle phases. (B) Fluorescence images of the ROS and GSH in different cell cycle phases which were characterized using two fluorescent probes after simultaneous staining. (C) Curves of fluorescence intensity of ROS variation in different cell cycle phases under dynamic and static conditions on a 1.5 mm microfluidic device. (D) Curves of fluorescence intensity of ROS variation in different cell cycle phases under dynamic and static conditions on a 5.0 mm microfluidic device. | ||
 | ||
| Fig. 6 Fluorescence intensity of intracellular GSH variation on the two kinds of microfluidic devices. (A) Curves of fluorescence intensity of GSH variation in different cell cycle phases under dynamic and static conditions on a 1.5 mm microfluidic device. (B) Curves of fluorescence intensity of GSH variation in different cell cycle phases under dynamic and static conditions on a 5.0 mm microfluidic device. | ||
In addition to the differentiation of cell cycle phases, a comparison between the dynamic and static conditions also shows that more severe QD cytotoxicity is observed under dynamic conditions at the same cell cycle phases. The QD concentration in channel 2 remains higher under dynamic conditions which could explain the results. The concentration difference between the two different long microchannels (Fig. 3A) also shows the same effect, the fluorescence intensity of the ROS is higher on the 1.5 mm microfluidic chip while the fluorescence intensity of GSH is lower (Fig. 5C and D and 6). The diffusion distance plays a certain role in QD cytotoxicity, which is in accordance with the reported results.35
Conclusions
In summary, a microfluidic device for single cell cycle observation was developed and the effect of cell cycle phases on QD cytotoxicity was demonstrated on this platform. QD accumulation in different phases ranked in the order G2/M > S > G0/G1 which led to the QD cytotoxicity of the cells in various phases also being ranked in the same order. The results implied that the cell cycle phases were also a considerable factor during the biological or toxicological study. The cell cycle must be accommodated in future studies and models to help understand the mechanism of QD cytotoxicity. It is noted that cancerous cells experience the S or G2/M phases more often than normal cells as they divide faster so the dilution of QDs is greater. The results imply that the imaging or therapy targeting of cancerous cells using QDs should combine strategies that one component slows or arrests cell division and another one implements the imaging or therapy. A comparison between the dynamic and static conditions and different diffusion distances were also conducted and the results agreed well with the previous report. The multi-functional microfluidic device is easily fabricated and could potentially be attached to high-throughput analysis systems for single cell research.Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos 91213305, 20935002, 21275088) and China Equipment and Education Resources System (no. CERS-1-75).Notes and references
- E. Pöselt, C. Schmidtke, S. Fischer, K. Peldschus, J. Salamon, H. Kloust, H. Tran, A. Pietsch, M. Heine, G. Adam, U. Schumacher, C. Wagener, S. Förster and H. Weller, ACS Nano, 2012, 6, 3346–3355 CrossRef PubMed.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS PubMed.
- M. Green, Angew. Chem., Int. Ed., 2004, 43, 4129–4131 CrossRef CAS PubMed.
- F. Song, P. S. Tang, H. Durst, D. T. Cramb and W. C. Chan, Angew. Chem., Int. Ed., 2012, 51, 8773–8777 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- W. E. Smith, J. Brownell, C. C. White, Z. Afsharinejad, J. Tsai, X. Hu, S. J. Polyak, X. Gao, T. J. Kavanagh and D. L. Eaton, ACS Nano, 2012, 6, 9475–9484 CrossRef CAS PubMed.
- N. Chen, Y. He, Y. Su, X. Li, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, Biomaterials, 2012, 33, 1238–1244 CrossRef CAS PubMed.
- C. Kirchner, T. Liedl, S. Kudera, T. Pellegrino, A. Muñoz Javier, H. E. Gaub, S. Stölzle, N. Fertig and W. J. Parak, Nano Lett., 2005, 5, 331–338 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- J. A. Kim, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 62–68 CrossRef CAS PubMed.
- B. Snijder, R. Sacher, P. Ramo, E. Damm, P. Liberali and L. Pelkmans, Nature, 2009, 461, 520–523 CrossRef CAS PubMed.
- A. Lesniak, A. Campbell, M. P. Monopoli, I. Lynch, A. Salvati and K. A. Dawson, Biomaterials, 2010, 31, 9511–9518 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Aberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- A. Nagy, J. A. Hollingsworth, B. Hu, A. Steinbruck, P. C. Stark, V. C. Rios, M. Vuyisich, M. H. Stewart, D. H. Atha, B. C. Nelson and R. Iyer, ACS Nano, 2013, 7, 8397–8411 CrossRef CAS PubMed.
- Y. Li, Y. Zhou, H. Wang, S. Perrett, Y. Zhao, Z. Tang and G. Nie, Angew. Chem., Int. Ed., 2011, 50, 5860–5864 CrossRef CAS PubMed.
- K. C. Nguyen, W. G. Willmore and A. F. Tayabali, Toxicology, 2013, 306, 114–123 CrossRef CAS PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS PubMed.
- V. Lecault, M. VanInsberghe, S. Sekulovic, D. J. H. F. Knapp, S. Wohrer, W. Bowden, F. Viel, T. McLaughlin, D. Falconnet and A. K. White, Nat. Methods, 2011, 8, 581–586 CrossRef CAS PubMed.
- P. Neuzi, S. Giselbrecht, K. Lange, T. J. Huang and A. Manz, Nat. Rev. Drug Discovery, 2012, 11, 620–632 CrossRef CAS PubMed.
- J. Wu, Q. Chen, W. Liu and J.-M. Lin, Lab Chip, 2013, 13, 1948–1954 RSC.
- A. M. Skelley, O. Kirak, H. Suh, R. Jaenisch and J. Voldman, Nat. Methods, 2009, 6, 147–152 CrossRef CAS PubMed.
- M. Kolnik, L. S. Tsimring and J. Hasty, Lab Chip, 2012, 12, 4732–4737 RSC.
- M. Morel, J. C. Galas, M. Dahan and V. Studer, Lab Chip, 2012, 12, 1340–1346 RSC.
- D. Bakstad, A. Adamson, D. G. Spiller and M. R. White, Curr. Opin. Biotechnol., 2012, 23, 103–109 CrossRef CAS PubMed.
- D. G. Spiller, C. D. Wood, D. A. Rand and M. R. H. White, Nature, 2010, 465, 736–745 CrossRef CAS PubMed.
- B. R. Singh, B. N. Singh, W. Khan, H. B. Singh and A. H. Naqvi, Biomaterials, 2012, 33, 5753–5767 CrossRef CAS PubMed.
- A. Nel, Science, 2005, 308, 804–806 CrossRef CAS PubMed.
- A. S. Thakor, R. Paulmurugan, P. Kempen, C. Zavaleta, R. Sinclair, T. F. Massoud and S. S. Gambhir, Small, 2011, 7, 126–136 CrossRef CAS PubMed.
- E. Boucrot and T. Kirchhausen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7939–7944 CrossRef CAS PubMed.
- J. K. Schweitzer, E. E. Burke, H. V. Goodson and C. D'Souza-Schorey, J. Biol. Chem., 2005, 280, 41628–41635 CrossRef CAS PubMed.
- D. Raucher and M. P. Sheetz, J. Cell Biol., 1999, 144, 497–506 CrossRef CAS.
- J. Wu, Q. Chen, W. Liu, Y. Zhang and J.-M. Lin, Lab Chip, 2012, 12, 3474–3480 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Single cell capture on the microfluidic device, concentration of the QD solution simulation in the microchannels and QD cytotoxicity detection using a CCK-8 assay kit. See DOI: 10.1039/c4ra01665c |
| This journal is © The Royal Society of Chemistry 2014 |