Programming galvanostatic rates for fast-charging lithium ion batteries: a graphite case
Younghoon Ko,
Yoon-Gyo Cho and
Hyun-Kon Song*
School of Energy and Chemical Engineering, UNIST, Ulsan 689-798, Korea. E-mail: philiphobi@hotmail.com
First published on 17th March 2014
Abstract
Galvanostatically induced lithiation of graphite, as a cathodic process of lithium ion batteries during charging, was investigated in situ by galvanostatic electrochemical impedance spectroscopy (GS-EIS). When lithiation is driven by charge rates slow enough for kinetics of the lithiation process to be considered relatively sluggish, charge transfer resistance (RCT) is slightly reduced as lithium ion intercalation proceeds from the dilute stage to stage 2L. Subsequently, RCT begins to increase during transformation of stage 2L to stage 2, followed by an abrupt increase in RCT observed during transition from stage 2 to stage 1, or after the inter-space of graphites is fully filled with lithium ions. As the ratio of charge rate to lithiated graphite increases, the potential responsible for the transition from stage 2L to stage 2 is shifted to more negative values due to significant polarization. Simultaneously, cells reach cut-off potentials before the transition from stage 2 to stage 1 proceeds. Based on the information regarding RCT profiles obtained by galvanostatic charging processes, a charging strategy is programmed with several different charge rates (C-rates). The capacity of lithiation is significantly enhanced by a C-rate switching (CRS) strategy. As a representative example, 75% of available capacity is charged for 50 minutes by a combination of 2 C, 1 C, and 0.5 C. However, only 12% and 51% of graphite is lithiated within the same time duration by a single charge rate of 0.1 C and 0.5 C, respectively.
1. Introduction
Lithium ion rechargeable batteries (LIBs) are expected to become the primary replacements for petroleum-based fuels in electric vehicles (EVs).1,2 LIBs provide a number of advantages in comparison with aqueous battery technologies, including higher energy density (100 to 170 W h kg−1 for LIBs versus ∼75 W h kg−1 for nickel–metal hydride batteries) and higher cell voltage (up to ∼4 V per cell).3,4 Carbonaceous materials, more specifically graphites, have been used predominantly as anode materials for LIBs. They are lithiated/delithiated reversibly with a significant amount of lithium (up to one lithium per six carbons) without any deterioration in their mechanical and electrical properties and their electrochemical potential value of Li+ intercalation is as low as that of metallic lithium. Additionally, they have well defined electrochemical characteristics which show several plateaus in chronopotentiomograms; this indicates higher capacity than other carbonaceous materials.5 However, the lithiation kinetics of graphites are not facile enough to satisfy the high power demand of EVs, even if they are superior to other alternatives such as conversion-reaction-based or alloying-based anode materials. Various efforts have been devoted to enhancing the kinetics of lithiation or delithiation of anode materials; as a pertinent example, a recent study reported that the lithiation process was kinetically improved by partially exfoliating edges of graphite.6Lithiation/delithiation kinetics are highly dependent on resistances associated with mass transfer and charge transfer processes. Lithium ions move through the electrolyte into the graphite surface (solution resistance, RS); penetrate the solid-electrolyte interface (SEI) layer RSEI; are intercalated from the SEI layer to the edge site of graphites (charge transfer resistance, RCT); and finally diffuse along the inter-space of graphites (Warburg impedance, ZW).7,8 The solid-state diffusion of Li+ (diffusion coefficient of Li+ in graphite, DLi+ = 10−12 to 10−8 cm2 s−1)9 can be the rate determining step at high rates of charging and discharging so that the charge transfer leading to LiC6 formation is limited. The concentration polarization, in addition to ohmic polarization, both induce higher overpotential during the charging of cells at high currents, causing the cell potential to reach a cut-off voltage before graphite is fully lithiated.
As graphite is lithiated with more amounts of Li+, electrochemical environments change so that the intercalation potential experienced by Li+ decreases. Step-wise potential changes have been observed depending on the periodicity of the lithiated layer.5,10 For example, the potential required for transforming one lithiated layer per four graphitic layers (stage 4) to stage 2 is higher than the potential required for transformation from stage 2 to stage 1. According to each stage (or the degree of lithiation within each stage), charge transfer kinetics vary with RCT as a function of potential. Useful information could be extracted from electrochemical impedance spectra if the lithiation of graphite during galvanostatic charging processes were monitored in real time using galvanostatic electrochemical impedance spectroscopy (GS-EIS). Typical impedance spectra have been obtained at a biased potential after electrochemical systems of interest are fully stabilized at the potential.9,11,12 With potentiostatic electrochemical impedance spectroscopy (PS-EIS), it is difficult to define the state of charge (SOC) at the potential at which impedances are obtained; this is because a wide range of SOC exists at a fixed potential, shown as potential plateaus over a time period in typical chronopotentiometric potential profiles. Another limitation of PS-EIS for investigating the lithiation of graphite anodes of LIBs is its inability to probe the rate-dependency of effective kinetic parameters. Impedances are obtained with PS-EIS after steady states (time-invariant conditions) are reached. This is not the case with GS-EIS, meaning that this technique may be used to investigate effective kinetic parameters and, thus, provide useful information about the galvanostatic lithiation of graphites. Related research has utilized non-stationary impedance analysis (equivalent to GS-EIS) based on impedance spectra as a function of time for investigating lead/acid batteries during galvanostatic charging.13,14 Additionally, the electrochemistry of electrodes for lithium batteries and LIBs was studied in situ by using GS-EIS to investigate the deposition/dissolution of the lithium metal anode15 and formation of the SEI layer on the graphite anode.10
Galvanostatic methods have been used primarily to charge or discharge practical electrochemical energy storage devices such as LIBs and supercapacitors. These methods control the rate of charging or discharging, thus, avoiding high current shocks that might be possible when using potential-controlled methods. Practically, a fixed value of current or C-rate (the ratio of current, with respect to the current charging or discharging energy at 100% level over a period of 1 hour), have been used without considering variable kinetics along electrochemical processes.
In this study, we adapted GS-EIS instrumentation by superimposing a biased current with a series of sinusoidal current waveforms. Kinetic parameters were extracted from coin half-cells based on graphite lithiation processes in real time or in situ by GS-EIS. The C-rate dependency of the kinetic parameters was also investigated. Based on the data arising from the study, we suggest a charging strategy, called the C-rate switching method (CRS), to enable a greater amount of energy to be stored within a fixed time period.
2. Experimental
2.1 Cell preparation
2032-type coin half-cells were configured with lithium foil as a counter electrode, with 1 M LiPF6 in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v ethylene carbonate–dimethyl carbonate (EC–DMC) as the electrolyte and a micro-porous polyethylene film (Tonen, 20 μm thick) as a separator. Natural graphite (NG; Mitsubishi Chemical, average particle size = ∼20 μm) was used as a working electrode, serving as the anode material of LIBs. The NG was coated on copper (19 μm thick) with 4% wt poly(vinylidene fluoride) (PVdF; Solet 5136) as a binder. Slurry of the NG and PVdF mixture in N-methyl-2-pyrrolidone was coated on copper foils, dried at 100 °C for 2 hours, and roll-pressed. Coin cells were assembled in an argon-filled glove-box with less than 1 ppm of oxygen and water. Assembled coin cells were kept at open circuit potential in a chamber at 25 °C (±1 °C) for 10 hours. Using a battery cycler (Wonatech/WBCS 3000), they, subsequently, were galvanostatically cycled twice between 1.5 and 0.01 V versus Li/Li+ at 0.1 C-rate to form the SEI layer and measure the capacity of each cell, respectively.
:5 v/v ethylene carbonate–dimethyl carbonate (EC–DMC) as the electrolyte and a micro-porous polyethylene film (Tonen, 20 μm thick) as a separator. Natural graphite (NG; Mitsubishi Chemical, average particle size = ∼20 μm) was used as a working electrode, serving as the anode material of LIBs. The NG was coated on copper (19 μm thick) with 4% wt poly(vinylidene fluoride) (PVdF; Solet 5136) as a binder. Slurry of the NG and PVdF mixture in N-methyl-2-pyrrolidone was coated on copper foils, dried at 100 °C for 2 hours, and roll-pressed. Coin cells were assembled in an argon-filled glove-box with less than 1 ppm of oxygen and water. Assembled coin cells were kept at open circuit potential in a chamber at 25 °C (±1 °C) for 10 hours. Using a battery cycler (Wonatech/WBCS 3000), they, subsequently, were galvanostatically cycled twice between 1.5 and 0.01 V versus Li/Li+ at 0.1 C-rate to form the SEI layer and measure the capacity of each cell, respectively.
2.2 Impedance measurement by GS-EIS
Impedances of the coin cells, experiencing two cycles of charging and discharging, were measured during galvanostatic lithiation by GS-EIS. Two channels of multichannel potentiostats (BioLogic/VSP-300) were interactively used (Fig. 1). Channel 1 applies a galvanostatic signal (a biased current) to the cells and measures resultant potential signals as a function of time. Channel 2 superimposes a series of small AC waves on the galvanostatic signal of Channel 1 and measures resultant potential waves to calculate impedances. Sinusoidal signals at 200 kHz to 1 Hz were used with 100 μA sinus amplitude. The sinusoidal current waves were applied to the constant current and the resultant potential waves were intermittently recorded, i.e. every 10 minutes for 0.05 C and 0.1 C; and every 5 minutes for 0.5 C and 1 C. Impedance data were fitted with a proposed equivalent circuit by using software (Princeton Applied Research, ZSimpWin). The best parameters were obtained by minimizing χ2. | ||
| Fig. 1 Inputs and outputs of GS-EIS. Input stimuli (c) are generated by superimposing small alternating current signals (b) on a galvanostatic signal (a). Temporal profiles of potential (d) and impedances (e) at points of interest (coloured circles in (d)) on the chronopotentiometric profile are measured simultaneously. | ||
2.3 C-rate switch (CRS) charging
All cells were kept potentiostatically on 1.5 V versus Li/Li+ at the battery cycler prior to charging. Three cells with identical fabrication and electrochemical history were used. The first and second cells as controls were lithiated galvanostatically at a fixed current of 0.1 C and 0.5 C, respectively. The third cell was lithiated with a series of C-rates: 2 C, 1 C and 0.5 C, consecutively. The C-rates were changed when the cells reached their cut-off potential at 0.01 C.3. Results and discussion
Lithium ions are intercalated into the interlayer space of graphite to form graphite intercalation compounds (GICs). The intercalation processes experience different electrochemical environments as they proceed. Lithium ions are intercalated, preferentially into the layers that are thermodynamically more favourable. To avoid the interference of one intercalated layer with another, periodicity of the intercalated layers is attained by holding a gap between them as a buffer. The next preferred interlayer space is filled with lithium ions after intercalation into the most preferred ones is completed. Each stage is identified with formation of its own GICs:5| LiC72 (dilute stage) ⇆ LiC36 (stage 4) | (I) |
| LiC36 (stage 4) ⇆ LiC27 (stage 3) ⇆ LiC18 (stage 2L) | (II) |
| LiC18 (stage 2L) ⇆ LiC12 (stage 2) | (III) |
| LiC12 (stage 2) ⇆ LiC6 (stage 1) | (IV) |
Chronopotentiometric profiles of lithiating natural graphite, at slow C-rates less than 0.1 C, showed several well-defined plateaus corresponding to each chemical equation shown above (Fig. 2a). Composition of GICs changes from the left-side to the right-side of the reactions as time or capacity increase at a fixed potential plateau. The staging behaviour becomes clearer in the dQ/dV plot obtained by differentiating capacity with respect to potential (Fig. 2b).
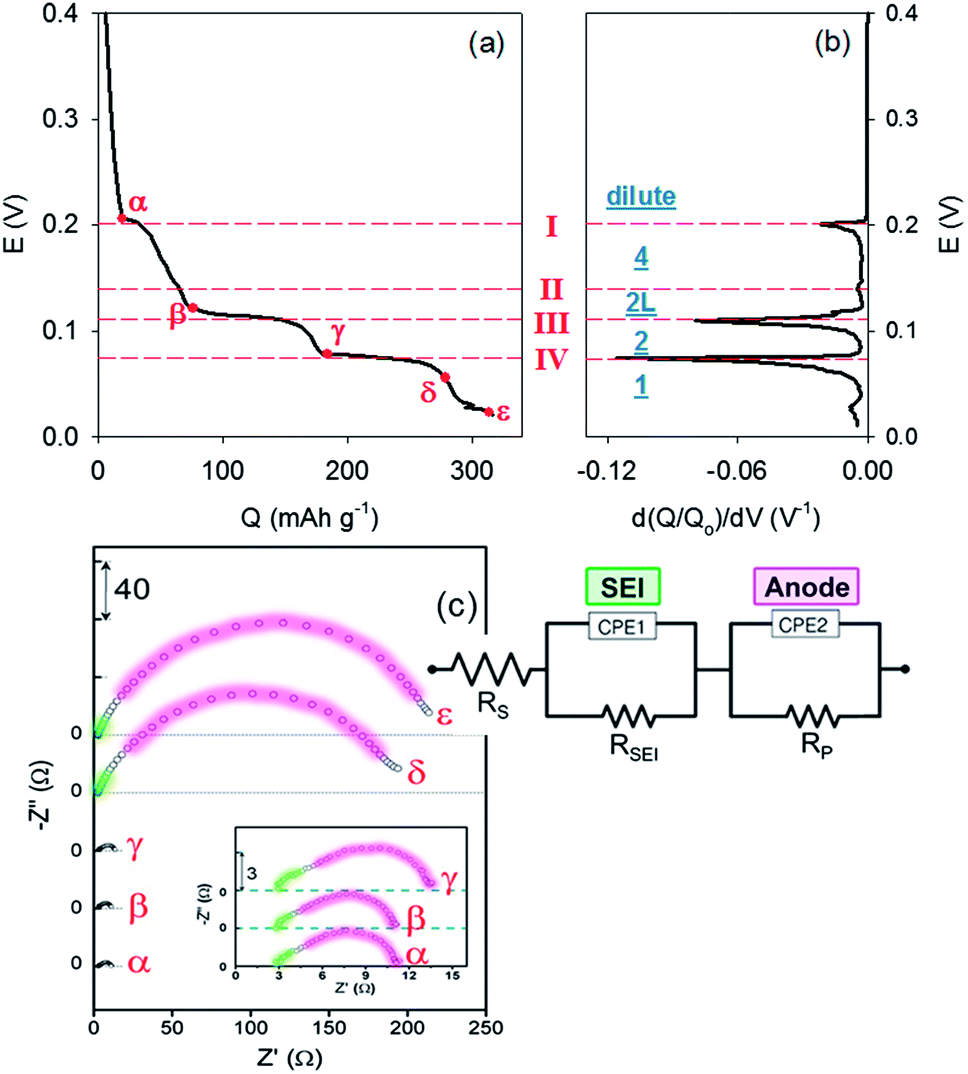 | ||
| Fig. 2 (a) Chronopotentiometric profile of lithiation of natural graphite at 0.05 C (Q = capacity; E = cell potential versus Li/Li+). (b) Differential capacity with respect to potential. Dashed red lines indicate peak potentials related to corresponding reactions (red Roman numbers). Underlined blue numbers or characters indicate stages of GICs. (c) Impedance spectra obtained at the points indicated by red Greek letters in (a). The impedance spectra at α, β and γ are magnified in the inset. An equivalent circuit was proposed to describe the impedance data. | ||
Impedance spectra were traced in situ during galvanostatic charging at 0.05 C. Representative impedance spectra are shown in Fig. 2c, which are measured at the red points designated by Greek letters in Fig. 2a. Two semi-circles, each of which is responsible for charge transfer through an interface, were highly overlapped with each other. The low-frequency semi-circle (red-coloured) was relatively larger than the high-frequency one (green-coloured). Charge transfers driven by Li+ through the first interface from the electrolyte to the solid-electrolyte interphase (SEI) and the second interface from the SEI to graphite cause the high-frequency and low-frequency semi-circles, respectively. These may be described by a combination of two Randles circuits (the equivalent circuit in Fig. 2c).16 Charge transfer resistance directly related to intercalation (RCT; diameter of the second semi-circle) dramatically increased at the end of reaction (IV), around 0.056 V, due to concentration polarization caused by saturated intercalation into graphite, leaving no availability for a concentration gradient to be developed. The high-frequency semi-circle became negligible compared with its low-frequency counterpart as GICs reached stage 1.
The values of RCT were estimated for the lithiation process at 0.05 C (black solid circles and lines of Fig. 3) after elements of the equivalent circuit were extracted from the impedance data (Fig. 2c) by nonlinear least square fitting. The capacity used as the abscissa was normalized by the capacity obtained at 0.05 C. Charge transfer resistances of impedance spectra obtained at higher rates (from 0.1 C to 1 C) are included in the same figure. When lithiation is driven by charge rates slow enough for kinetics of the lithiation process to be considered relatively sluggish, RCT was slightly reduced with lithium ion intercalation proceeding from the dilute stage (α); to stage 4; and then to stage 2L (β) (eqn (I) and (II)). Values of RCT then began to increase from the formation of stage 2L to stage 2 (γ) (III), followed by an abrupt increase in RCT observed during transition from stage 2 to stage 1 (δ) (IV) or after the inter-space of graphites is fully filled with lithium ions.
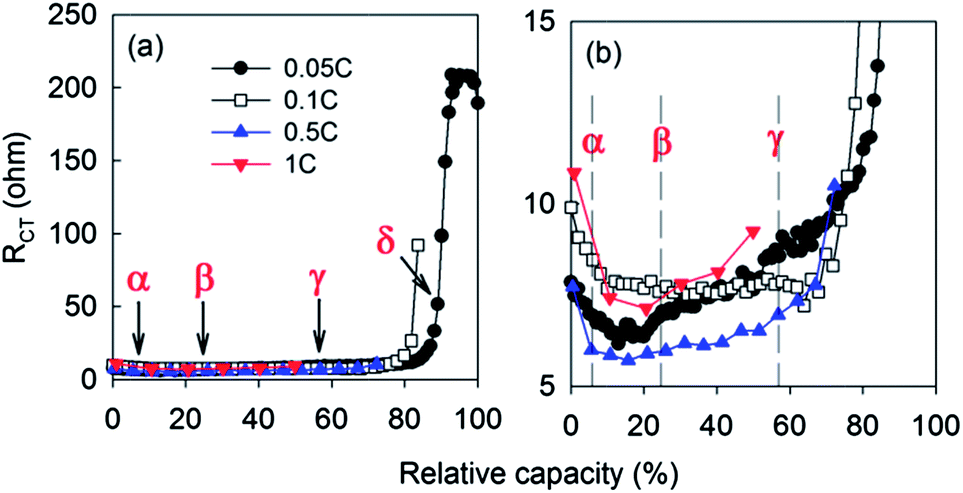 | ||
| Fig. 3 (a) Change of RCT along lithiation process at various C-rates. (b) Magnified plots of (a). | ||
The decrease in RCT during GIC transition from the dilute stage to stage 2L can be understood in terms of the interlayer distances (d) of graphites. The values of d were reported at 0.335 nm for bare graphite (or very dilute stage); 2.718 nm for stage 8 (between the dilute stage and stage 4); 1.376 nm for stage 4; and 0.704 nm for stage 2.5,17 The increase in d spacing possibly leads to reduced hindrance against introducing lithium ions into the interlayer space and enhanced solid-state diffusion within graphite.
As the ratio of charge rate to lithiated graphite increases, the onset capacity of abrupt increase in RCT is shifted to smaller values as shown clearly from the comparison of RCT between 0.05 C and 0.1 C (open squares in Fig. 3a). The RCT curves at higher rates (0.5 C and 1 C) are expected to go up earlier than that of 0.1 C (Fig. 3b) even if they reached the cut-off voltage before showing the abrupt increase. The effect of C-rate was investigated in a more detailed way by comparing the RCT profiles with differential capacity (dQ/dV) (Fig. 4). Development of overpotential with increasing rate is clearly shown by the shift of peak potentials of GIC transitions in a negative direction (blue and green solid lines). Independent of C-rates, the potentials at which RCT is minimized corresponded to GIC transition (II) from stage 4 to stage 2L. In other words, electrochemical factors that favourably affect the kinetics of lithiation are improved up to stage 2L, while the stages from 2 to 1 provide electrochemical environments unfavourable to lithiation processes.
 | ||
| Fig. 4 Capacity differentiated with respect to potential (dQ/dV, black curve for left ordinate) and charge transfer resistance (RCT, red curve for right ordinate) as a function of potential (E). These curves were obtained simultaneously from chronopotentiometry at three different C-rates: (a) 0.05 C, (b) 0.5 C and (c) 1 C. | ||
At this point, the usefulness of our GS-EIS method should be emphasized. We measured impedances under lithiating graphites at constant current. Our GS-EIS method effectively takes a “snapshot” of the dynamic states or stages during charging, which is the most important aspect of distinguishing GS-EIS and conventional PS-EIS. In PS-EIS, as a traditional method of EIS, impedances are measured in a stationary and equilibrated state after systems are stabilized under an applied constant potential.18 PS-EIS releases electrochemical information only applicable to the systems controlled by very small C-rates. Even in this case, we are unsure how the information could be used if there is a hysteresis between charging and discharging profiles. In particular, under high applied currents, electrochemical parameters obtained from the equilibrated state cannot describe electrochemical behaviours of dynamic states. The impedance measured by our GS-EIS (Fig. 3 and 4) shows its raison d'être: impedance spectra depend on C-rates.
Based on what we have learned from our GS-EIS studies (i.e. that resistance related to charge transfer decreases up to stage 2L, then increases abruptly and exponentially), a question may be raised: what if charging current is programmed to minimize the effects of RCT for lithiating graphite or cells based on a graphite anode? Use of high currents at stage 2 to stage 1 should be avoided due to their high RCT values.
A simple sequence of three different C-rates (2 C–1 C–0.5 C) was programmed to lithiate graphite more efficiently and rapidly. Higher currents were used at less resistive stages and vice versa. Use of a programmed sequence is not the best solution, but was adequate for testing the feasibility of our C-rate switching strategy. The most optimized profile could be obtained by considering RCT profiles at various C-rates and their effects on lithiation kinetics which could form the basis for further studies. The CRS strategy was compared with lithiation at fixed rates at 0.1 C or 0.5 C in terms of capacity reached within a fixed time span (50 minutes) (Fig. 5). It is obvious that slower charging accommodates higher capacity only if the charging time is not the concern. However, fast charging is one of the most important issues as LIB-based electric vehicles are developed and commercialized. Within 50 minutes, 12% and 51% of available capacity were charged at 0.1 C and 0.5 C, respectively. There was no significant difference of potential profiles between those C-rates because impedance relevant to stages of up to 50% relative capacity is kept at a low level. Therefore, there were no reasons not to initially use higher rates. As the first step of the CRS sequence, fast lithiation at 2 C delivered 18% of available capacity. Subsequent lithiation at 1 C and 0.5 C provided an additional 25% and 32% capacity, respectively. As a result, the CRS strategy enabled cells to be efficiently charged at capacities 7 and 1.5 times as high as that obtained at 0.1 C and 0.5 C, respectively.
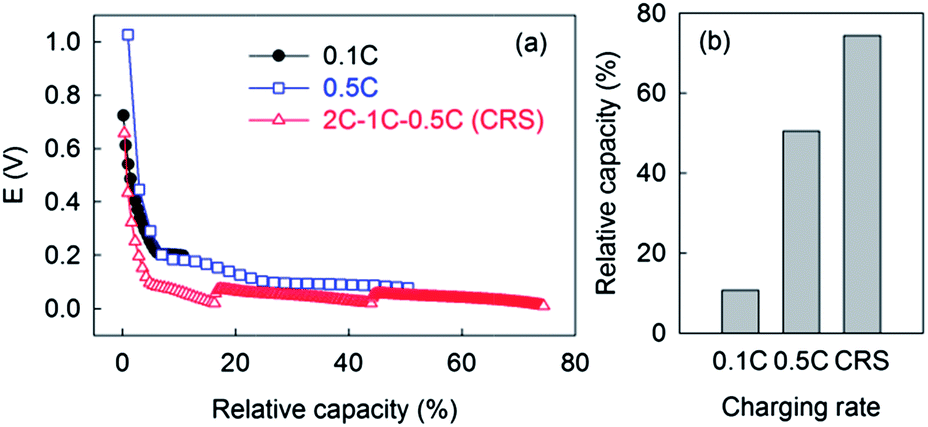 | ||
| Fig. 5 Lithiation of graphite by C-rate switching strategy versus conventional fixed rate strategy. (a) Potential profiles. (b) Relative capacity of lithiation. | ||
Lithiation into graphite was described schematically at a fixed rate (0.1 C) in comparison to our CRS strategy in Fig. 6. Solvated lithium ions are introduced into the space between graphitic layers after passing through a solid-electrolyte interphase (SEI) layer.8,19,20 The SEI layer is an ionic conductor but an electrical insulator. When lithium ions pass through the SEI layer, they experience resistance (RSEI in Fig. 2c) that is independent of the stages of GICs. However, RCT relevant to Li+ introduction from the SEI layer to graphite depends strongly on the stages. The intercalated Li+ diffuses through the interlayer space, leaving room for subsequent lithium ions.9,21 At a slower fixed charging rate (0.1 C, left column of Fig. 6), Li+ introduction into the interlayer space of graphite is slower than Li+ movement within the space. That is to say, lithiation is not limited by the solid-state diffusion. However, it takes a greater period of time to charge cells because the slow rate was used. In comparison, at high charging rates (2 C in the right column of Fig. 6), intercalated lithium ions accumulate at the entrance of the interlayer space (around edges of graphitic layers) due to limited mass transfer within graphite. In our CRS strategy, the traffic of Li+ movement is mitigated by changing the C-rate into slower ones. Additionally, the increase in RCT,caused by a high degree of lithiation, is compensated by adopting a sequence of decreasing C-rates as discussed above.
 | ||
| Fig. 6 Lithiation of graphite at 0.1 C (left column) compared to our CRS strategy (right column). | ||
4. Conclusions
The use of a sequence of different C-rates, instead of conventional constant current charging, was proposed for fast charging of LIBs. The CRS strategy was devised based on information regarding the lithiation of graphite investigated in situ by GS-EIS. Our GS-EIS method successfully identified meaningful kinetic parameters of graphite lithiation in a non-stationary and non-equilibrated state during chronopotentiometric charging at a fixed current or C-rate. Charge transfer resistance was minimized at GIC transition (II) from stage 4 to stage 2L, followed by a mild increase of RCT over stage 2L and 2, and an abrupt increase after GIC transition (IV) from stage 2 to stage 1. C-rate dependency of kinetic parameters was also identified, which cannot be established by conventional PS-EIS. Based on the kinetic behaviour of graphite lithiation, a sequence of C-rates for charging cells was constructed with decreasing C-rates (2 C–1 C–0.5 C) to avoid the abrupt development of resistive environments during the latter part of the lithiation process. Within a fixed time (50 minutes), seven-fold and one-and-a-half-fold capacities were reached by our CRS charging, in comparison with the values obtained by conventional charging at 0.1 C and 0.5 C, respectively.Acknowledgements
This work was supported by MOTIE (Green: 10042948 (KEIT), Star: 20135020900030), MSIP (Mid: 2013R1A2A2A04015706 (NRF), CRC: 2013K000210) and MOE (BK21Plus: 10Z20130011057), Korea.Notes and references
- J. Li, B. L. Armstrong, J. Kiggans, C. Daniel and D. L. Wood, J. Electrochem. Soc., 2013, 160, A201–A206 CrossRef CAS PubMed.
- T. H. Kim, J. S. Park, S. K. Chang, S. Choi, J. H. Ryu and H. K. Song, Adv. Energy Mater., 2012, 2, 860–872 CrossRef CAS PubMed.
- D. Linden and T. B. Reddy, Handbook of batteries, Mc Graw-Hill, New York, 2nd edn, 2002 Search PubMed.
- V. Srinivasan, AIP Conf. Proc., 2008, 1044, 283–296 CrossRef CAS PubMed.
- T. Ohzuku, Y. Iwakoshi and K. Sawai, J. Electrochem. Soc., 1993, 140, 2490–2498 CrossRef CAS PubMed.
- J.-S. Park, M.-H. Lee, I.-Y. Jeon, H.-S. Park, J.-B. Baek and H.-K. Song, ACS Nano, 2012, 6, 10770–10775 CAS.
- Y. C. Chang, J. H. Jong and G. T. K. Fey, J. Electrochem. Soc., 2000, 147, 2033–2038 CrossRef CAS PubMed.
- T. Abe, H. Fukuda, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1120–A1123 CrossRef CAS PubMed.
- T. Piao, S. M. Park, C. H. Doh and S. I. Moon, J. Electrochem. Soc., 1999, 146, 2794–2798 CrossRef CAS PubMed.
- M. Itagaki, N. Kobari, S. Yotsuda, K. Watanabe, S. Kinoshita and M. Ue, J. Power Sources, 2004, 135, 255–261 CrossRef CAS PubMed.
- C. Wang, A. J. Appleby and F. E. Little, Electrochim. Acta, 2001, 46, 1793–1813 CrossRef CAS.
- X.-G. Sun and S. Dai, J. Power Sources, 2010, 195, 4266–4271 CrossRef CAS PubMed.
- Z. Stoynov and B. Savova-Stoynov, J. Electroanal. Chem., 1985, 183, 133–144 CrossRef.
- Z. Stoynov, B. Savova-Stoynov and T. Kossev, J. Power Sources, 1990, 30, 275–285 CrossRef.
- T. Osaka, T. Momma and T. Tajima, Denki Kagaku oyobi Kogyo Butsuri Kagaku, 1994, 62, 350–351 CAS.
- A. Funabiki, M. Inaba, T. Abe and Z. Ogumi, J. Electrochem. Soc., 1999, 146, 2443–2448 CrossRef CAS PubMed.
- K. Xu and A. von Cresce, J. Mater. Chem., 2011, 21, 9849–9864 RSC.
- Y. Ko and S.-M. Park, J. Phys. Chem. C, 2012, 116, 7260–7268 CAS.
- J. Fischer, Chemical physics of intercalation, Plenum Press, New York, 1987, p. 59–78 Search PubMed.
- A. Funabiki, M. Inaba, Z. Ogumi, S. i. Yuasa, J. Otsuji and A. Tasaka, J. Electrochem. Soc., 1998, 145, 172–178 CrossRef CAS PubMed.
- V. A. Sethuraman, L. J. Hardwick, V. Srinivasan and R. Kostecki, J. Power Sources, 2010, 195, 3655–3660 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |