Microfluidic long-term differential oxygenation for bacterial growth characteristics analyses†
Xin Cui,
Hon Ming Yip,
Qian Zhu,
Chengpeng Yang and
Raymond H. W. Lam*
Department of Mechanical and Biomedical Engineering, City University of Hong Kong, Hong Kong. E-mail: rhwlam@cityu.edu.hk; Fax: +852-3442-0172; Tel: +852-3442-8577
First published on 25th March 2014
Abstract
Dissolved oxygen is a critical micro-environmental factor to determine the growth characteristics of bacteria, such as cell viability, migration, aggregation and metabolic processes. Here, we report a microfluidic long-term oxygenator which can support high-throughput parallel cell cultures under multiple defined oxygenation conditions (0–42 ppm). An array of oxygen–nitrogen gas micro-mixers is developed and fabricated in the device to generate stable oxygen concentrations for the oxygenation process. A water-jacket layer located between the gas layer and the cell culture chamber is applied to block evaporation and maintain the conditions of the culture media in the chamber. Furthermore, we perform simulations and experiments to investigate the functions of the gas mixers and the water jackets. We also conduct culture experiments to study the long-term growth (up to one week) and aggregation of three selected dental bacteria (Streptococcus mutans, Actinomyces viscosus and Fusobacterium nucleatum) under ranges of oxygenation conditions. These particular results can provide important insights into their roles in dental biofilm formation. Overall, this work demonstrates that the long-term microfluidic oxygenation approach can effectively regulate defined dissolved-oxygen levels in cell microenvironments. Importantly, this oxygenation approach can be further applied to general long-term analyses of cells for their behavioral, metabolic and genetic responses, and their biofilm formation processes.
Introduction
Dissolved oxygen (DO) plays an important role in bacterial cell growth behaviors, including cell viability,1,2 migration,3–5 and metabolic processes in biological systems.6–8 Oxygen molecules function as electron acceptors or oxidizing agents involved in many aerobic metabolic activities of bacterial species9,10. On the other hand, extremely high oxygen concentrations can be toxic for both aerobic and anaerobic species because toxic forms of oxygen (e.g. superoxide radical and hydrogen peroxide) are generated.9,10 The ranges of DO levels for favorable bacterial cell growths are species-specific. For example, Desulfovibrio vulgar as obligate anaerobic bacteria prefers DO concentrations ranging from 0.02 to 0.04% (or from 0.24 to 0.48 μM); and excessive DO (>0.08%) can restrain the bacteria growth.3,11 For facultative anaerobic bacteria such as Escherichia coli can change their metabolism schemes from aerobic to anaerobic when there is insufficient DO or nutrient concentrations in the microenvironment.12,13 Oxygen concentrations suppress the gene expressions of ArcB (sensor) and ArcA (transcriptional regulator) which are responsible for aerobic metabolism in E. coli.14 Therefore, systems supporting parallel incubation of cells under different DO concentrations are requisite in order to perform more comprehensive investigations for bacterial behaviors under different oxygen microenvironments and the role of DO in specific bacteria.Microfluidics has been proven as an extremely effective technology for cell biology study, mainly due to its wide range of advantages over traditional macro-scale cell culture methods, e.g. high throughput analysis,15,16 shorter response time,17,18 automated sample operations, and reduced consumption of cells and reagents.15,19 In particular, microfluidics can achieve sample transport, manipulation and processing20–22 in the micro-meter scale, and therefore the local biological microenvironments such as DO levels can be regulated and mimicked relatively easily.18,23–25 Up to date, several approaches have been reported to generate different DO levels in multiple microchannels or microchambers for cell culture applications. Many of these microfluidic devices were made of polydimethylsiloxane (PDMS) mainly due to its material properties such as optically transparency, gas permeability, biocompatibility and fabrication feasibility based on soft lithography.26,27 Park et al. used different microelectrodes in underneath microchannels to obtain different oxygen production rates based on water electrolysis.28 Due to the diffusivity of oxygen through the membrane between a microchannel and an oxygen generation region, additional oxygen concentrations could be generated and diffuse into the microchannel. Though this method had a high oxygen generation rate, the fabrication of microelectrodes was relatively complicated and the precise DO level was hard to maintain. A small amount of hydrogen peroxide, which was harmful to cells, could be produced in the water electrolysis process.29 Recently, Chen et al. developed strategies to apply reactions to generate oxygen.7 They designed two channels for oxygen generation and oxygen removal on the both sides of a cell culture microchannel. This method required precise controls of flow rates of different chemical substances to maintain a specified DO concentration in the culture channel. Hydrogen peroxide was also involved in the oxygen generation. Droplets-based methods were implemented to improve efficiency of oxygen diffusion in microchannels, in which carrier oil functioned as a gas source of a predefined oxygen concentration;30 thereby DO in water droplets quickly equilibrated with that in the surrounding oil. However, this approach cannot be applied in long-term cell culture because of the limited nutrients in water droplets. A widely adopted strategy for oxygen concentration modulation was the two-layer microchannel structure containing a layer of gas mixing channels fabricated directly overhead the culture channels, separated by a gas-permeable PDMS membrane for the oxygen diffusion.31–33 Due to the small thickness of the diffusion membrane, oxygen with defined concentrations diffused quickly into the underneath microchannels. Because the DO level in media was determined by the ratio of oxygen along the gas channel, the DO level was stable under a fixed condition along the gas channel.
Although the two-layer structure provided stable oxygenation without the disadvantages of other mentioned approaches, it faced a technical challenge of preventing evaporation of culture media because the convective flow along overhead the gas channels continuously brought away water molecules evaporating through the diffusion membrane.34–37 Such moisture loss over time, even at a relatively small rate, reduced the relative osmolarity in culture media, resulting in increase of cell stress and even cell death.37,38 Although the moisture loss could be reduced by applying pre-humidification of the gases,39–41 experiments showed that overhead gas flow still induced significant moisture evaporation. Our previous work introduced pre-humidified oxygen and nitrogen into a differential oxygen concentration generator, by flowing the required gases (nitrogen and oxygen) through water reservoirs upstream of the device inlets. This approach could lengthen the cell culture period up to <10 h.42 The oxygenation approach is yet to be improved in order to support further investigations on long-term cell behaviors such as cell aggregation, biofilm formation, and mild cytotoxicity responses, which develop only after days or weeks of culture.43–45
In this work, we demonstrate a microfluidic strategy to implement long-term oxygenation and precisely define different oxygen concentrations in multiple culture chambers in a microfluidic device. We configure the differential oxygenator as a series of parallel gas mixing channels. A target oxygen concentration can be defined precisely in each mixer by regulating the flow resistances in the inlets channels for pure oxygen and pure nitrogen. This type of oxygenator has more robust performance than many other oxygen gradient generators.6,8,42,46–48 Further, in order to support the long-term cell culture, a water jacket layer is fabricated between the gas layer and the cell culture layer. This water jacket acts as a sacrificial layer of water molecules for blocking the evaporation of culture media via the gas-permeable PDMS. In the oxygenated culture experiments, Streptococcus mutans, Actinomyces viscosus and Fusobacterium nucleatum are selected due to their different oxygen concentration requirements and their critical roles in the dental biofilm formation. S. mutans assist growth of many other dental bacterial species in human oral cavities; and they likely cause the dental caries formation.49–51 A. viscosus bacteria have been proven by experiments for their capability of causing root surface caries and periodontal bone lesions.52,53 The anaerobic F. nucleatum can bridge aerobic bacteria and anaerobic bacteria to grow together in dental biofilms. Without F. nucleatum, most anaerobic oral species cannot survive in the oral cavity under sufficient oxygen concentrations.54–56 Over the cell culture periods (up to 3–7 days), we monitor the cell densities and distributions in each culture chamber. We investigate the optimal oxygen levels for growths of selected bacteria types, and the effects of oxygen levels on the bacteria growths and auto-aggregation capabilities.
Methods and materials
Device fabrication
Multilayer soft lithography was applied to fabricate the microfluidic long-term oxygenator containing a gas layer, a water jacket layer, a control layer and a flow layer, as shown in Fig. 1. The molds for all these layers were manufactured by photolithography. For the gas layer, the water jacket layer and the control layer, microstructures on the molds were patterned with SU-8 negative photoresist on silicon wafers with thickness of 100 μm, 20 μm, and 20 μm, respectively. To fabricate a mold with a thickness of 20 μm, SU-8 photoresist (SU-8 10, Microchem, Newton, MA) was first spin-coated (SM-180 Easy, SAWATEC AG, Hong Kong) on a silicon wafer. Then it was baked at 65 °C for 2 min and baked again at 95 °C for 5 min on a hotplate. After ultraviolet (UV) exposure (350 nm, 10 mW cm−2, 40 s) with a precise mask film, post-exposure bake was conducted at 65 °C for 1 min and then at 95 °C for 2 min. After the exposed photoresist was developed with the SU-8 developer (Microchem), the mold was placed again on a hotplate at 95 °C for 1 min. For the gas-layer mold, after spin-coating photoresist (SU-8 50, Microchem, Newton, MA) for a thickness of 100 μm on a silicon wafer, soft bake was conducted by baking the wafer on a hotplate at 65 °C for 10 min and baked again at 95 °C for 30 min. Then the photoresist was exposed under UV light (350 nm, 10 mW cm−2, 100 s) for the desired patterns, followed by post-exposure bake at 65 °C for 1 min and then at 95 °C for 10 min. After developing the photoresist, the mold was baked again at 95 °C for 1 min in order to further cross link the photoresist.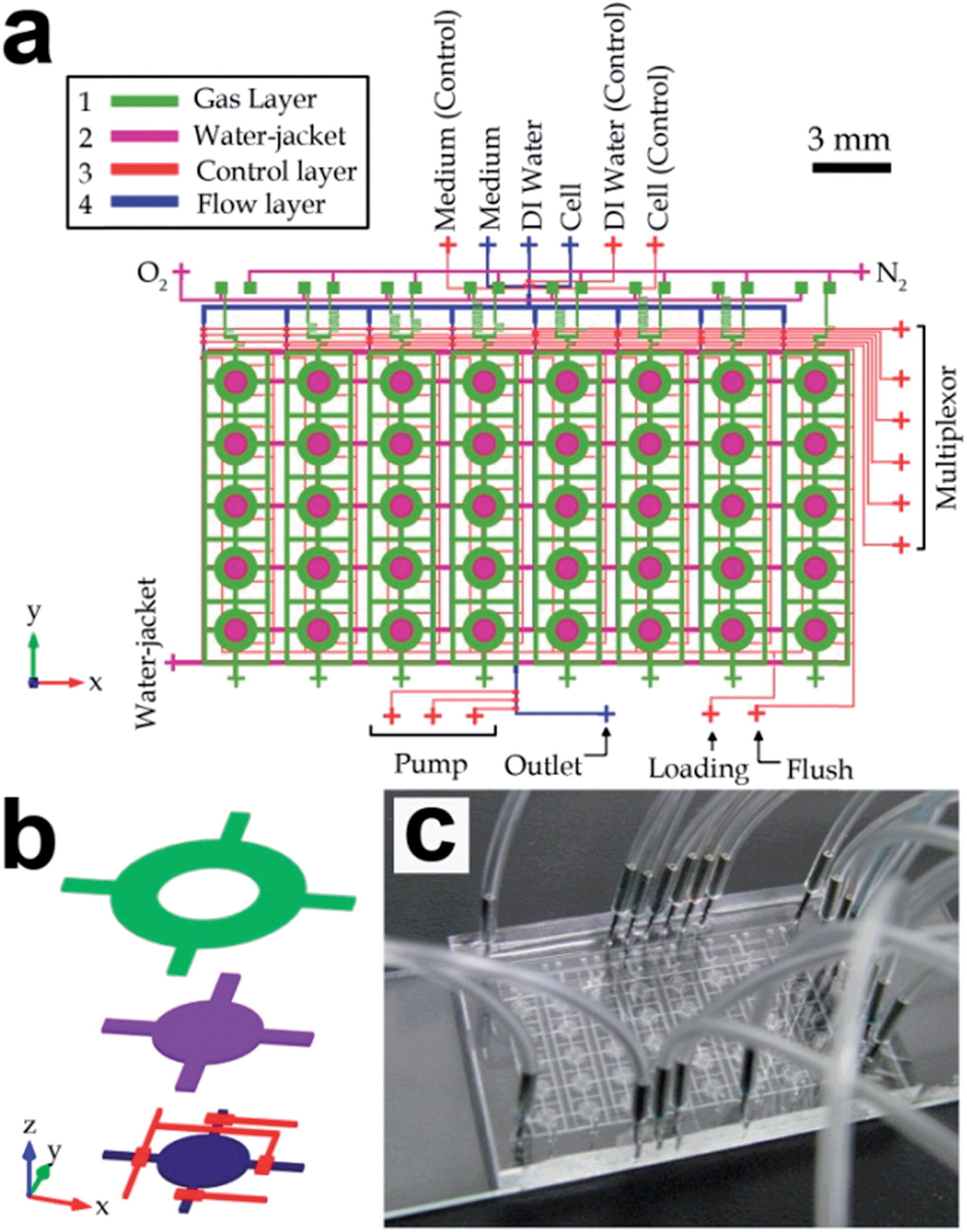 | ||
| Fig. 1 Microfluidic long-term oxygenated cell culture device containing 5 × 8 culture chambers. (a) Device layout, which consists of four layers of channel structures, i.e. a gas layer, a water-jacket layer, a control layer and a flow layer fabricated from top to bottom. (b) Three-dimensional structure of an individual cell culture region (spacing between layers is not in scale). (c) Photograph of a fabricated device. | ||
Mold structures for the flow layer was fabricated with a positive photoresist (AZ4620, AZ Electronic Materials, Branchburg, NJ) in order to obtain the required channel geometry for microfluidic pneumatic valves and peristaltic pumps. Before spin-coating the AZ photoresist on a silicon wafer for a thickness of 20 μm, hexamethyldisilazane (Sigma-Aldrich, St. Louis, MO) was applied on the wafer to promote the adhesion between the photoresist and the silicon wafer. Then the wafer was baked on a hotplate for 30 min and exposed under UV (350 nm, 10 mW cm−2, 80 s). Afterwards, the mold was developed in the AZ 400K developer (AZ Electronic Materials, Branchburg, NJ) and reflowed on a hotplate at 140 °C for 1 min to achieve the round-shaped channel profiles.
Before fabricating the multilayer PDMS device, all the molds should be first silanized with a high-molecular-weight trichloro-perfluorooctyl saline (Sigma-Aldrich) to facilitate the polydimethylsiloxane (PDMS) release from the molds during the subsequent device fabrication processes. PDMS monomer and the curing agent (Sylgard 184, Dow Corning, Midland, Michigan) were mixed with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio and degassed in a vacuum bell jar. The top device layer with gas channel structures was fabricated by pouring the PDMS pre-polymer onto gas channel mold fixed in a Petri dish for a thickness of ∼3 mm, baking the sample in an oven at 80 °C for 20 min, and peeling off the PDMS substrate part from the mold. Holes for oxygen and nitrogen inlets were punched using a Harris punch (Harris Uni-Core, TedPella, Redding, CA) with a diameter of 0.5 mm.
:1 weight ratio and degassed in a vacuum bell jar. The top device layer with gas channel structures was fabricated by pouring the PDMS pre-polymer onto gas channel mold fixed in a Petri dish for a thickness of ∼3 mm, baking the sample in an oven at 80 °C for 20 min, and peeling off the PDMS substrate part from the mold. Holes for oxygen and nitrogen inlets were punched using a Harris punch (Harris Uni-Core, TedPella, Redding, CA) with a diameter of 0.5 mm.
Based on multilayer soft lithography, stacked PDMS layers including the water jacket, the control layer and the flow layer were sequentially fabricated with thicknesses of 60 μm, 300 μm and 40 μm, respectively. In brief, PDMS pre-polymer was spin-coated on the second mold with a thickness of 60 μm and baked in an oven at 80 °C for 14 min. After alignment under a stereo microscope (JSZ-6S, Yee Mau Industrial Co., Hong Kong), the first layer of PDMS with punched holes was bonded with the second layer and baked again at 80 °C for 2 h. Then, the two-layer PDMS substrate was peeled off from the silicon mold, followed by punching holes for the gas outlets. Through-holes between the gas inlet channels and the parallel gas mixer structures were cut using a razor blade (Bard-Parker stainless steel surgical blade, Becton Dickinson and Company) at the overlapping regions (yellow squares in Fig. 2c). With the similar soft lithography procedures, each of the control layer and the flow layer was fabricated sequentially by spin-coating with PDMS on the mold, baking at 80 °C in an oven until partial cure, aligning with the PDMS substrate under a stereo microscope, further baking at 80 °C for 2 h, peeling off from the mold, and punching holes for fluidic connections. Afterwards, the four-layer PDMS should be treated by oxygen plasma (PDC-002, Harrick Plasma, New York) with 29.6 W power for 1 min in order to bond the stacked PDMS onto a glass slide (75 mm (l) × 25 mm (w) × 1 mm (h); Cat # 16004-422, VWR).
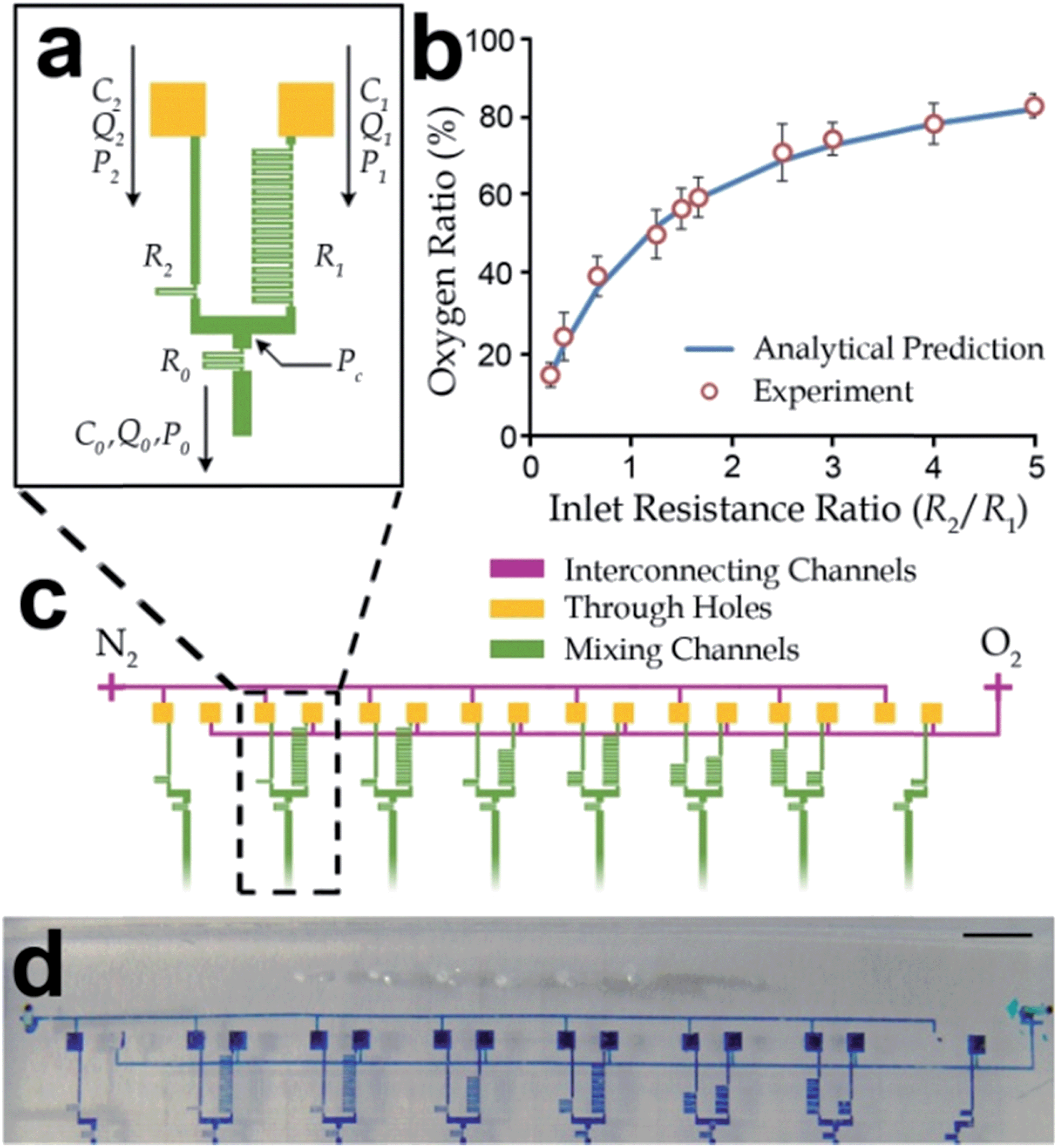 | ||
| Fig. 2 Design of parallel gas mixers for differential oxygenation. (a) Layout of a gas mixer with two gas inlets and one gas outlet. The outlet oxygen concentration can be regulated by defining different fluidic resistances of the inlet channels (R1 and R2). (b) Experimental results of oxygen concentrations at the outlets of the gas mixers with different ratios of fluidic resistances (R1/R2). Each value and the corresponding standard deviation (error bar) were obtained from at least three independent experiments. (c) The differential oxygenator contains three layers of microchannel structures with an upper flow layer for oxygen and nitrogen gases, through-holes in the lower layer for fluidic connections, and eight pairs of gas mixers to provide different defined oxygen concentration at their outlets. (d) Micrograph of the differential oxygenation fabricated in the microfluidic long-term oxygenator. A color dye was injected along the gas channels for better visualization of the channel structures. Scale bar: 2 mm. | ||
Bacteria cell preparation
Bacteria species Streptococcus mutans, Actinomyces viscosus and Fusobacterium nucleatum have been selected to study their long-term growth under various oxygen concentrations. Traditional cell cultivation was performed to maintain and expand the cell populations. A. viscosus (ATCC 19246) bacteria were cultured in the Brain Heart Infusion (BHI) broth (Hardy Diagnostics R20) in an incubator with a rotary shaking platform (200 rpm) for 20 h at 37 °C. S. mutans (ATCC 25175) bacteria were also cultured in the above conditions but without the rotary shaking platform. F. nucleatum (ATCC 10953) bacteria were cultured in a GasPak anaerobic jar (BD 26026) within Pre-Reduced and Anaerobically Sterilized (PRAS) chopped meat media (Hardy Diagnostics AS 811). The required anaerobic environment was maintained using gas-generating sachets (BD 260678) in the anaerobic jar.Oxygenated cell culture
Each fabricated microfluidic oxygenator was sterilized by baking at 100 °C for ∼10 h and exposed them under UV light for ∼2 h. The microfluidic flow channels in the device were rinsed with 1× phosphate-buffered saline (PBS), pH 7.4. Occasionally, some channel regions might contain air bubbles, which were then removed by pressurizing the PBS buffer with additional air pressure from the channel inlets. Air trapped in the flow channels could escape through the gas-permeable PDMS substrates. On the other hand, we injected distilled water and culture media into the water jacket layer and the flow layer, respectively, with a gauge pressure of 3 psi at the corresponding inlets. All the valves in the control layer were closed with a gas pressure of 12 psi in order to maintain water in closed cavities until cells were injected later. We pre-conditioned temperature and gas concentration of the device by placing the device at 37 °C in an incubator and simultaneously supplying pre-humidified oxygen and nitrogen gases to the oxygen concentration gradient generator in the gas layer. Prior to the oxygenated cell culture, the chamber walls in the device were pretreated with human saliva for >30 min to form glycoprotein-rich pellicle layers and facilitate cell attachments. The human saliva was extracted from a 30 year-old healthy male and was pre-filtered by a syringe filter (pore size: 0.22 μm. EMD Millipore, Billerica, MA) to remove all the bacteria.We extracted each bacteria sample from the bulk culture and diluted to a targeted seeding density (106–107 cells per ml). We then injected the bacteria sample into the microfluidic oxygenator with a syringe pump (PicoPlus, Harvard Apparatus, Holliston, MA) at a flow rate of 0.05 μl min−1. During the injection process, a microfluidic multiplexor in the device opened up each column of microchambers in the device consecutively. We placed the device in a temperature-controlled water bath at 37 °C for oxygenated cell culture. The cells were incubated under a static condition (without liquid flows) for ∼2 h in order to enable the cell attachment in the saliva-coated culture chamber. Afterwards, nitrogen and oxygen gas sources for the gas mixing were maintained at a constant pressure (1 kPa), meanwhile fresh culture media were periodically flowed into each column of chambers for 10 s every 1 h by a syringe pump (PicoPlus, Harvard Apparatus) at a flow rate of 0.003 μl min−1, until the experiments ended. At different time points during the cell culture, we captured phase-contrast microscopic images at all culture chambers and analyzed for the oxygenated cell growth using a customized Matlab (Mathworks, Natick, Massachusetts) program.
Image processing
We developed a programming script in Matlab to process micrographs of cells in each culture chamber at different time points during experiments. Bacterial cells appeared as dark spots in a phase-contrast microscopic image focused properly. We applied a Gaussian mask (mask size: 50 pixel × 50 pixel, Gaussian radius: 10 pixel) for low-pass image filtering, in order to estimate any uneven exposure of the microscope light source. We then subtracted intensities of the raw micrograph by the Gaussian-filtered image; and therefore the uneven background intensities were effectively eliminated. We implemented thresholding with a cutoff intensity (a value of 12 out of 256 levels) on the processed image to extract the bacterial communities (i.e. the darker spots) and to further quantify the corresponding cell surface coverage in the chamber area.Results and discussion
Microfluidic long-term oxygenator design
In this work, we developed a microfluidic device consisting of multi-layers of microchannels to precisely generate different DO concentrations for long-term cell culture. The layout of the device is shown in Fig. 1a & b and a fabricated device is shown in Fig. 1c. This device contained an array of forty (five rows and eight columns) identical functional units for high-throughput cell analyses. For each unit (Fig. 1b), the uppermost gas channel delivered gases with a defined oxygen ratio generated by parallel gas mixers (described in the next paragraph). The water-jacket layer below functioned as a shield layer to block any transfer of moisture between the gas channel and the other channels/chamber structures underneath this water jacket. The water jacket was located above each culture chambers. All the water-jacket structures were interconnected together such that only one inlet was only required for the water insertion. The underneath microchannel/chambers structures included control valves, and a culture chamber connecting with other flow channels located at the lowest layer of channel structures. Together, oxygenation of culture media was achieved by oxygen diffusion across the separating PDMS membranes and the water jacket. On the other hand, we considered that microchannels flowed with the bacteria samples during the cell seeding process could be contaminated; and hence different flow channels were used for cell seeding and media insertion. For each culture chamber (Fig. 1a & b), the ‘vertical’ channels (y-direction) cross each culture chamber were assigned for cell seeding, whereas the ‘horizontal’ channels (x-direction) were flowed with fresh media to maintain the cell growth in the chamber.Furthermore, eight gas mixers were fabricated in the gas layer to generate eight independent oxygen concentrations. Each of the mixers consisted of a gas inlet for oxygen, a gas inlet for nitrogen and a gas outlet for the mixed gas with specified oxygen volumetric concentration as shown in Fig. 2a. Because of the fact that all the gas inlets were connected to the common gas sources (i.e. oxygen and nitrogen gas tanks), we adopted a two-layer microchannel structure, in which two inter-connecting microchannels were fabricated in the secondary layer to route all mixer inlets for the same gas type (oxygen or nitrogen) to a common gas port of the device. As shown in Fig. 2c, connections of the gas channels were achieved by cutting through-holes (yellow squares) at the overlapping channel patterns between the two inter-connecting gas lines (purple) and the eight mixers (green). In addition, we located the inter-connecting gas lines in the same layer with the water jackets, in order to eliminate an extra PDMS layer of the fabrication process (Fig. 1a). Each mixed gas with a defined oxygen level will flow along a downstream gas channel, fabricated overhead a water jacket and a column of five culture chambers.
Other than control valves, a microfluidic multiplexor16 was designed in the control layer to selectively enable flows along one column (out of eight columns) of five cell culture chambers. This configuration facilitated accessibility of the cell seeding and the medium replacement processes during the long-term cell culture. Considering also the arrangement of the overhead gas channels, it provided an identical microenvironment in the five chambers for repeating culture experiments. Overall, this device can support high-throughput long-term oxygenated cell culture for the growth analysis with eight independent DO concentrations in each column of the five identical cell culture chambers.
Gas mixer design
As mentioned, the gases mixed with different volumetric ratios of oxygen and nitrogen were generated by eight gas mixers in the device. Comparing to the commonly used cascade mixer array,32,42,57 the configuration of parallel gas mixers should have a more robust performance. It is because any defect/blockage along any single mixing channel in the cascade mixer array can affect the mixed concentrations at all the outlets, while a defect along a mixing channel in a gas mixer will affect only the corresponding mixing unit.To generalize our oxygenator design, we modeled a gas mixer with cases for different flow resistances of the two upstream channels as illustrated in Fig. 2a. These flow resistances and hence the flow rates of oxygen and nitrogen were controlled by different lengths of the upstream folded channels. In addition, we set the channel dimensions of the downstream mixing folded channel such that the representative scaled Peclet number (Pe*) for the possible range of gas flow rates (10−7–10−6 m3 s−1) was sufficiently small, i.e. Pe* = 10−3–10−1, as shown in Table 1. The scaled Peclet number is defined as Pe* ≈ UW/D × W/L, where L, W, and H are channel length, width and height, respectively; U is the average gas flow velocity (U = Q/(WH)); and D is the diffusivity of oxygen in nitrogen. In our analyses, L, W, H and U should have different values in different channel sections. This condition implied diffusion dominated over the convective effect along the mixing channel. Oxygen and nitrogen were then fully mixed along the downstream channel with a concentration defined by the ratio of the gas flow rates.
| Folded channel | Length L (μm) | WidthW (μm) | Height H (μm) | Flow rate (Q) × 10−6 m3 s−1 | Resistance × 108 (Ns m−5) | Re* × 10−3 | Pe* × 10−2 |
|---|---|---|---|---|---|---|---|
| a Note: scaled Reynolds number: Re* ≈ ρUL/μ × H2/L2, where U = Q/(WH) is the average velocity; scaled Peclet number: Pe* ≈ UW/D × W/L, where D is 2.19 × 10−4 m2 s−1. | |||||||
| Oxygen inlet | 0–129600 |
28 | 20 | 0–1.43 | 0–252.14 | 0.42–16.7 | 0.28–11.38 |
| Nitrogen inlet | 0–9000 | 28 | 20 | 0–3.55 | 0–15.76 | 4.39–41.36 | 3.44–32.43 |
| Outlet | 1800 | 28 | 20 | 1.16–3.75 | 0.64–3.5 | 13.48–43.71 | 9.99–34.02 |
The corresponding oxygen concentration at the mixer outlet Co (0 ≤ Co ≤ 1) can be estimated based on the conservation of mass,
| Co = Q1/(Q1 + Q2), | (1) |
To estimate the channel resistance, we have configured the channel dimensions such that the scaled Reynolds numbers (Re*) for all channel sections in the mixer were in a range of 10−3–10−1. Viscous effects dominated in all the channels and the velocity profiles of the gas flows were fully developed. The scaled Reynolds number is defined as Re* ≈ ρUL/μ × H2/L2, where ρ is the density of gas mixture; μ is viscosity of the gas flowing in the channel. It should be mentioned that viscosity of the gas mixture along the mixing channel can be estimated by
| μ ≈ 1/(C1/μO2 + C2/μN2), | (2) |
The fluidic resistance for a flow channel with a rectangular cross-section can then be expressed as
 | (3) |
We performed experiments to validate the microfluidic gas mixer for different ratios of flow resistances between the oxygen and nitrogen inlet channels. The channel dimensions and operation parameters are shown in Table 1. Pure oxygen and nitrogen gas tanks were used as gas sources at a common gauge pressure of 1 kPa. The emission intensities from PtOEPK were measurement with multiple oxygen sensing modules we developed previously.42 We connected an oxygen sensing channel embedded with platinum(II) octaethylporphine ketone (PtOEPK) to every outlet from the gas mixers. Briefly, the measurement mechanism of oxygen concentration was based on the change in emission intensity from a PtOEPK sensor under an external excitation light exposure; and the emission intensity reflected the oxygen content in a gas mixture.42 Experimental results (Fig. 2b) show that oxygen concentrations from the device gas outlets for different ratios of flow resistances along gas inlet channels have very good agreements with the analytical values calculated by the method mentioned above. This relation implies that oxygen concentrations can be effective tuned by the dimensions of inlet gas channels in the microfluidic parallel gas mixers as shown in Fig. 2d.
Oxygenation
It has been proven previously that DO concentrations in a culture chamber can be effectively regulated by an overhead channel flowing gas mixtures with defined oxygen ratios.42 For the long-term oxygenator, the PDMS membranes between the gas and liquid layers facilitate the gas diffusion into the culture chambers, yet the membrane thickness is constrained by addition of the water-jacket layer. Moreover, a PDMS membrane between the culture chamber and the water jacket should be sufficiently thick (∼300 μm for the devices in this work) to provide adequate mechanical supports against deformations caused by hydraulic pressures from the water jacket or the culture chamber. We configured the overhead gas perfusion channel as ring-shaped chamber such that the further PDMS deflections caused by the supplying gas pressure can be avoided.Here, we describe the characterization of the oxygenation process in the long-term oxygenator, by simulating oxygen concentration profiles over the cross sections of individual culture regions configured with different combinations of the design parameters. We applied commercial software (COMSOL Multiphysics 4.2, Burlington, MA) to perform these simulations on a 2D axisymmetric model of an individual culture region (Fig. 3a), which was composed of a cylindrical culture chamber (radius: 500 μm; height: 20 μm), a cylindrical water-jacket chamber (radius: 600 μm; height: 20 μm), and a ring-shaped overhead gas channel. The simulations were based on modifying the three design parameters of the ring-shaped gas perfusion channel from a reference model: (1) height, (2) inner radius and (3) width of the ring-shaped gas perfusion channel. The thicknesses of PDMS layers on the upper and lower sides of the water jacket were 40 μm and 300 μm, respectively. The oxygen profiles for different device configurations were calculated by solving the diffusion equations. Diffusivities of oxygen in PDMS and in water were set as 5.0 × 10−5 cm2 s−1 and 2.0 × 10−5 cm2 s−1, respectively. We considered a normalized oxygen concentration with 100% as the imposed oxygen level in the ring-shaped gas channel and 0% as the oxygen level in the ambient environment. For the boundary conditions, a constant oxygen level (100%) was defined at the boundaries of the ring-shaped gas channel. A zero flux was set at the bottom side of the model because oxygen could not diffuse into the underneath glass slide. In addition, oxygen concentration at the upper boundary of the model was set as 0%. The oxygen concentration profile at the bottom of the culture chamber for each configuration modified from the reference model was considered, because this region represented the cell microenvironment more directly. Effects of the three varying design parameters of the ring-shaped gas perfusion channel: height (10–200 μm), inner radius (0–1000 μm) and width (100–1000 μm), modified from the reference model are shown in Fig. 3b–d, respectively. In summary, these results show that >95% of the oxygen concentration in gas chamber can be regulated at the bottom of the culture chamber for the current microfluidic long-term oxygenator (height: 100 μm; inner radius: 550 μm; width: 500 μm).
 | ||
| Fig. 3 Parametric simulation study on the microfluidic oxygenation. (a) Numerical models were established with the same geometric configurations as the fabricated devices. The DO profiles at the bottom of the culture chamber (diameter: 500 μm) as functions of (b) the height, (c) the inner radius and (d) width of the ring-shaped gas perfusion channels. Note that the symbols R, H and W only refer to this figure. | ||
Humidity control using a water jacket
A water-jacket layer was embedded between the gas layer and the culture layer in the device, in order to minimize evaporation of culture media and eliminate osmolality changes of the media and the resultant cell death.37,38 The water jacket functioned as a sacrificial layer of media evaporation. We conducted numerical simulations using COMSOL 4.2 to investigate the humidity regulation in culture chambers using the water jackets. The axisymmetric simulation model (Fig. 4a) depicted the cross-sections of the humidity levels and gradients on the chamber regions with and without a water jacket. The model dimensions were set identical to those of the fabricated long-term oxygenators. Because the diffusivity of oxygen in water (2.0 × 10−5 cm2 s−1) is comparable to that of oxygen in PDMS, the addition of water jacket layer between gas layer and culture layer should not induce a significant effect on oxygenation of the culture chambers. The moisture levels at the boundaries of the water jacket and the culture chamber were both predefined to be 100% because these chambers will be filled with water and medium in cell culture process. The humidity levels at the boundaries of ring-shaped gas microchannel and of the upper device boundary were set to be 0% because we would like to consider the worst scenarios. The flux at the lower device boundary was set to be zero as there was no diffusion of water/moisture into the glass substrate. Fig. 4a & b shows the profiles of humidity levels and gradients for the cases with and a water-jacket layer. We considered that humidity gradients around the chambers represented the fluxes of moisture across the chamber boundaries based on the Fick's law of diffusion. The simulated humidity gradients along the upper and side chamber boundaries are described in Fig. 4b. This result indicates that the water jacket layer can effectively reduce the loss of moisture due to the convective evaporation via the overhead gas flow by 75.87% compared with the case without the water jacket. | ||
| Fig. 4 Humidity regulation of culture chambers by adding a water jacket layer between the gas and flow layers. (a) Simulated axisymmetric humidity profiles of a culture region with a water jacket (right) and without a water jacket (left). (b) Simulated axisymmetric humidity gradient profiles of a culture region with a water jacket (right) and without a water jacket (left). (c) Comparisons of humidity gradients between the cases of the microchambers with and without a water jacket in the device. Profiles of the humidity gradients on the chamber top (left) and the chamber side wall (right) are plotted here. (d) Representative images of a liquid residual in a micro-chamber at different times (0 h, 2 h, 2.5 h and 3 h) throughout evaporation. The liquid areas (enclosed by dotted-line boundaries) were used to quantify the evaporation rates. This experiment was performed for an equivalent device with humidified gas and a water-jacket layer. Scale bar: 500 μm. (e) Results of three sets of water evaporation experiments. Gas with humidification was achieved by flowing gas through deionized water. The error bars were obtained from data of three independent experiments. Statistical significance is shown by the p-values (< 0.05) obtained by the Student's t-test. | ||
We also performed experiments to validate the evaporation reduction of the water jacket layer embedded in the device. The experiments consisted of four cases: (1) applying dry compressed air as the gas sources for both mixer inlets to a device without a water-jacket layer (the PDMS between the chamber and gas layers was then ∼360 μm thick), (2) applying pre-humidified air to mixers without a water-jacket layer, (3) applying dry air with an embedded water jacket, and (4) applying pre-humidified air to mixers with a water-jacket layer. In these experiments, we considered one chamber in the microfluidic oxygenator because configurations of the chambers are identical. We performed at least three sets of independent experiments for each case in order to obtain the average evaporation processes. To begin the experiments, we injected water in every culture chamber and isolated the area by closing the two valves around the chamber, followed by application of the compressed dry air flow. The gas pre-humidification was achieved by passing the air into a bottle of distilled water before the air flowing to the device inlets. The evaporation process in the chamber was recorded by taking time-lapse images under an inverted microscope. Throughout the experiments, an air bubble formed and expanded in the chamber. The bubble formation was due to the gas permeability of PDMS, i.e. external air replaced the evaporated water in the chamber by diffusing via the PDMS substrate of the device. The bubble formation indicated the volumetric reduction of water due to evaporation, as shown in Fig. 4d. Results (see Fig. 4e and S2†) show that the evaporation duration for the case with a water jacket was much longer than the cases without a water jacket. We can observe also that using pre-humidified air (instead of dry air) induced a slower evaporation process. Indeed, applying both the gases with pre-humidification and a water-jacket layer in the oxygenator can further eliminate the medium evaporation in the microchambers for the long-term cell culture applications.
Long-term bacteria growth under different dissolved oxygen levels
We performed experiments to investigate the long-term (up to about one week) bacteria growth under different levels or oxygenation. Three bacterial species commonly found in human oral cavities (Streptococcus mutans, Actinomyces viscosus and Fusobacterium nucleatum) were selected due to their important roles on the dental biofilm formation. We configured channel dimensions in the eight gas mixers of the device to generate oxygen volumetric ratios (0–100%) in order to investigate the corresponding bacteria growths under different DO levels, ranging from 0 ppm (obligate anaerobic) to 42 ppm (fully oxygenated). Specifically, we set the DO for the aerobic F. nucleatum with denser levels in the anaerobic range (0–8 ppm), considering that the bacteria should be very sensitive to the aerobic microenvironment. During the oxygenated cell culture, microscopic images were taken at each cell culture chamber every 1–2 days. Cell growth images for each column of culture chambers were processed by a customized MATLAB script as described in Methods and materials. Fig. 5 contains the representative processed images and raw images for the three selected dental bacteria. The processed images effectively show the cell densities and the cell distributions for the bacterial species extracted by the image processing scheme. The cell distribution in each chamber after the cell injection and attachment initiation steps (cell densities for all cell types were ∼1 × 107 cells per ml) had a high uniformity. After seven days of culture, significant increments of the bacteria populations were observed in the chambers; and the levels of increments depended of both the bacteria type and the DO levels (see Fig. 6 and S1†). For comparisons, we normalized total cell densities by scaling the values to ‘1’ as the initial cell densities measured right after cell seeding. It should be noted that the measured increments in the cell densities may not accurately describe the cell growth rates in the exponential phase because parts of the suspending cells have been flushed away by the medium flows. Nevertheless, these observed increments in cell densities can sufficiently reflect a favorable DO level for the bacteria growth. It has been clearly shown that facultative anaerobic S. mutans and A. viscosus had higher growth rates under aerobic environments. S. mutans and A. viscosus obtained maximum growth rates under ∼16 ppm and ∼11 ppm DO levels, respectively. For F. nucleatum, a maximum growth rate is shown under an absolutely anaerobic condition (0 ppm). | ||
| Fig. 5 Image processing of the cell distributions after one week of cell culture for the three selected bacterial species. S. mutans and A. viscosus, grew under an aerobic condition (DO ∼ 8 ppm), whereas F. nucleatum grew under an anaerobic environment (DO ∼ 0 ppm) in the long-term oxygenator. Only the regions in the chambers labeled by dotted-line circles in the raw images were considered for image processing and analysis. Scale bar: 250 μm. | ||
 | ||
| Fig. 6 Cell populations of three bacterial species (S. mutans, A. viscosus and F. nucleatum) after one week of oxygenated cell culture. (a) Representative cell distributions (after the image processing) of S. mutans, A. viscosus and F. nucleatum under eight selected dissolved oxygen levels. The scale bars are 100 μm. (b) Cell densities of the three bacteria as functions of dissolved oxygen levels and culture duration. Each value of the cell densities in the charts represents the average of five independent experiments; and it was normalized with the cell density as ‘1’ at the beginning of culture (0 h). | ||
As shown in Fig. 6b, the facultative anaerobic bacteria S. mutans grew in both aerobic and anaerobic environments. Our experimental results have shown also oxygen-tolerance of S. mutans under relatively high DO levels (>8 ppm). This cell behavior was consistent with the previous research.49,58 Nevertheless, the cell growth was not as rapid under a nearly oxygen-saturated environment. Generally, hydrogen peroxide produced by Streptococci in the over-oxygenated (>8 ppm) microenvironment can be a factor to inhibit bacterial growth, resulting from the lack of catalase.49,58 It has been reported that exposure of hydrogen peroxide, especially for longer durations (>10 h), can lead to permanent cell damage and therefore retardation of growth.59 Another possible cause is that oxygen may change gene expressions of bacteria in their long-term growth (>20 h), causing alterations of the energy-generating pathways for carbohydrate metabolism. It has been proven that the oxygen exposure can down-regulate the enzymes involved in production of adhesive glucan exopolysaccharides, hence cell attachment and growth in the culture chambers should be inhibited accordingly.51 Results of A. viscosus show a similar growth trend with S. mutans as A. viscosus is also an facultative anaerobic species.60 Previous research has reported that A. viscosus can alter its metabolic mechanisms to adapt different oxygen levels in their growth environments.60 In addition, F. nucleatum is known to be the strictly anaerobic species,55,56 therefore it can only grow under very low DO levels (up to ∼6 ppm), as also indicated in our experiments. We can also observe in Fig. 6b that under aerobic microenvironments, F. nucleatum still exhibits cell growth, although the growth is very slow. Indeed, it has been reported that cell–cell interactions involved in the cell clusters may enhance the oxygen-tolerance of the entire population.56,61
Aggregation of the selected bacteria
The bacterial aggregation should partially depend on the cell density in a community.25,62–64 If the cell density is extremely low, seldom bacteria can express strong aggregation abilities. Furthermore, it has been reported that certain genes with abilities of regulating intracellular signals for aggregation are likely to be expressed under specified ranges of oxygen concentrations,51,65–67 resulting in their great aggregation abilities under only specific DO environments. For instance, the previous study on aggregation abilities of F. nucleatum revealed the auto-aggregation could determine a large number of gene expressions and stimulate different metabolic processes, such as the tyrosine phenol-lyases and serine-related metabolisms.55 Though many previous research works focused on aggregation among different oral bacterial species,53,54,56 the bacterial aggregation abilities under different DO concentrations are yet to be studied quantitatively.Considering that the bacterial growth is highly sensitive to DO as demonstrated in our previous experiments, the DO concentration may affect significantly the selected bacteria in this work on their auto-aggregation abilities, as a cell response independent from the cell growth rate. During the cell culture up to several days, the cells in the culture chambers would grow and aggregate simultaneously, mainly initiated by the intercellular communications including migration and cell–cell attachment.62 Therefore, a highly aggregating bacterial species should form bigger cell clusters in the cell populations; whereas a species without noticeable aggregation capability should distribute sparsely while its cell population expands.
Aggregation abilities of the three selected bacterial species (S. mutans, A. viscosus and F. nucleatum) were examined for oxygenations of different DO levels (0–42 ppm). Similar to the cell growth experiments described in the previous section, the cells were first seeded into the saliva-coated chambers. Typically, previous research examined the increments of sizes of cell clusters to quantify the cell aggregation abilities.53,54,68 Based on the same principle, we adopted the aggregation rate, which is defined as the relative increment of cluster area per day, to evaluate the cell aggregation abilities under different DO levels. Image processing was applied to identify the cell clusters in each culture chambers. Results for S. mutans indicate significant auto-aggregation rates under relatively low (≤10 ppm) DO concentrations (Fig. 7, left). For the growth under higher DO levels, S. mutans have expressed low or even negative auto-aggregation rates. This behavior can be explained by the possible harmful effects of the produced hydrogen peroxide59 and the down-regulations of the cell adhesion-related enzyme generation processes51 under an over-oxygenated environment, as mentioned in the previous section. Results for A. viscosus (Fig. 7, middle) have shown the significant aggregation ability under all the oxygenation levels, but there was no clear trend between the oxygen level, cell growth and the aggregation rate. To our best knowledge, the auto-aggregation ability of A. viscosus has not yet been quantitatively characterized in the previous research. On the other hand, F. nucleatum bacteria showed their aggregation abilities with a correlation to the growth under different DO conditions (Fig. 6b, right; and Fig. 7, right). F. nucleatum showed relatively greater auto-aggregation ability compared with S. mutans, as also reported in other related research.54,55 Therefore, it may be concluded that the auto-aggregation of bacteria under different DO microenvironments is highly independent of the cell growth rate, or at a minimum such behavior is species-specific.
 | ||
| Fig. 7 Aggregation abilities of S. mutans, A. viscosus and F. nucleatum under different DO levels. The aggregation rates were defined by the relative increment of coverage for each cell colonies over one-day growth. The error bars represent standard errors calculated from five independent culture experiments. | ||
Furthermore, the aggregation of multiple bacterial species in a dental biofilm offers the fundamental cell–cell interactions during the biofilm formation. It has been well proven that a bacterial species can aggregate with multiple species, because the species may have different recognition sites (e.g. polysaccharides56) for attachment of other bacterial species. The aggregation provides also preferred microenvironments for multiple species growing simultaneously in the biofilm.54,56 S. mutans, A. viscosus and F. nucleatum have important and irreplaceable roles in formation of the dental biofilm, in which the bacterial aggregation affects a wide range of genes expressions, stimulates metabolic pathways for energy sources, and eventually diversifies the cellular metabolism schemes.55 In the early stage of dental biofilm formation, some bacteria species (mainly Streptococci including S. mutans) adhere to pellicles on tooth surfaces and initiate the attachment of other species throughout the formation processes.49–51 One of these species is Actinomyces sp., such as A. viscosus, that has a great adhesion capability in biofilm formation.69,70 The interaction between Streptococci and Actinomyces forms the primary community of dental biofilm.71 The aggregation ability of F. nucleatum is particularly important in the dental biofilm formation. F. nucleatum is known as a ‘bridging’ species as it can also co-aggregate with most aerobic and anaerobic bacteria species (up to hundreds of known cell types56,72–74), subsequently colonizing and forming microflora as mature dental biofilms in human oral cavities.54–56 Interestingly, as shown in this work, although the growth of S. mutans and A. viscosus are restrained in anaerobic environment, the aggregation abilities for the three bacteria (including also F. nucleatum) are all significant. This observation has a good agreement with our understanding on dental biofilm formation that the bacteria aggregation often occurs more rapidly in a closed oral cavity, in which the DO level is relatively low after the oxygen consumption by the aerobic dental bacteria. This quantified bacteria behavior may provide further insights to the dental biofilm formation and further effective ways to inhibit the formation process.
Conclusion
A microfluidic long-term device has been developed for parallel oxygenation of multiple defined dissolved oxygen (DO) levels (0–42 ppm) to support the high-throughput bacteria growth analyses in this research. We reported for the first an effective oxygenation approach to maintain long-term bacteria growth up to about one week. The series of gas mixers were configured as parallel mixing units such that the mixing ratios between nitrogen and oxygen were adjusted by the channel dimensions in the mixers. We fabricated culture chambers underneath the gas mixing layer to facilitate oxygenation of culture media in the chambers by gas diffusion via the separating gas permeable membrane structures. Furthermore, an additional water-jacket layer was embedded between the gas channels and the culture chambers in the device. In this case, culture media evaporation in the chambers, which was promoted mainly by the convective flow along the overhead gas channel, was shielded by the water jacket for the ∼75.87% reduction of water loss. We performed simulations and experiments to characterize the device dimensions for the optimal oxygenation efficiency (>95%).We applied the oxygenated culture to study the growth characteristics of three dental bacteria species: S. mutans, A. viscosus and F. nucleatum. Our results showed that their growth rates and aggregation capabilities were dependent on the cell type and highly sensitive to the DO levels; and more importantly, the bacterial aggregation was independent of their growths. Interestingly, all the three chosen bacteria showed significant aggregation capabilities in the anaerobic microenvironment. These results may provide important insights to deepen our understanding of the biofilm formation mechanism. In essence, this work has demonstrated that an effective and stable oxygenation strategy, which can provide a defined DO environment for long-term analyses of cell growth and biofilm formation. This strategy can be further applied to general high-throughput studies on cell and biofilm behaviors responding to DO levels, such as aggregations and cell–cell communications.
Acknowledgements
We sincerely thank supports for the early development of this research from Department of Mechanical Engineering in Massachusetts Institute of Technology. We appreciate the invaluable advices from Dr T. Thorsen. We acknowledge the financial supports from City University of Hong Kong (project# 7200267 and 9610212), Croucher Foundation Scholarship and Early Career Scheme of Hong Kong Research Grant Council (project# RGC124212).References
- A. Oller, C. Buser, M. Tyo and W. Thilly, J. Cell Sci., 1989, 94, 43–49 Search PubMed.
- M. Radisic, J. Malda, E. Epping, W. Geng, R. Langer and G. Vunjak-Novakovic, Biotechnol. Bioeng., 2006, 93, 332–343 CrossRef CAS PubMed.
- A. Eschemann, M. Kühl and H. Cypionka, Environ. Microbiol., 1999, 1, 489–494 CrossRef CAS.
- R. Grishanin and S. Bibikov, Biosci. Rep., 1997, 17, 77–83 CrossRef CAS.
- B. L. Taylor, I. B. Zhulin and M. S. Johnson, Annu. Rev. Microbiol., 1999, 53, 103–128 CrossRef CAS PubMed.
- M. Adler, M. Erickstad, E. Gutierrez and A. Groisman, Lab Chip, 2012, 12, 4835–4847 RSC.
- Y.-A. Chen, A. D. King, H.-C. Shih, C.-C. Peng, C.-Y. Wu, W.-H. Liao and Y.-C. Tung, Lab Chip, 2011, 11, 3626–3633 RSC.
- J. F. Lo, E. Sinkala and D. T. Eddington, Lab Chip, 2010, 10, 2394–2401 RSC.
- P. S. Stewart, J. Bacteriol., 2003, 185, 1485–1491 CrossRef CAS.
- I. Zhulin, M. Johnson and B. Taylor, Biosci. Rep., 1997, 17, 335–342 CrossRef CAS.
- H. Cypionka, Annu. Rev. Microbiol., 2000, 54, 827–848 CrossRef CAS PubMed.
- E. M. Gregory and I. Fridovich, J. Bacteriol., 1973, 114, 1193–1197 CAS.
- L. S. Wong, M. S. Johnson, I. B. Zhulin and B. L. Taylor, J. Bacteriol., 1995, 177, 3985–3991 CAS.
- G. Unden, S. Becker, J. Bongaerts, J. Schirawski and S. Six, Antonie van Leeuwenhoek, 1994, 66, 3–22 CrossRef CAS.
- J. Melin and S. R. Quake, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 213–231 CrossRef CAS PubMed.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS PubMed.
- D. J. Beebe, G. A. Mensing and G. M. Walker, Annu. Rev. Biomed. Eng., 2002, 4, 261–286 CrossRef CAS PubMed.
- C. Yi, C.-W. Li, S. Ji and M. Yang, Anal. Chim. Acta, 2006, 560, 1–23 CrossRef CAS PubMed.
- V. Srinivasan, V. K. Pamula and R. B. Fair, Lab Chip, 2004, 4, 310–315 RSC.
- P. Gravesen, J. Branebjerg and O. S. Jensen, J. Micromech. Microeng., 1993, 3, 168 CrossRef CAS.
- T. M. Squires and S. R. Quake, Rev. Mod. Phys., 2005, 77, 977 CrossRef CAS.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- E. Brouzes, M. Medkova, N. Savenelli, D. Marran, M. Twardowski, J. B. Hutchison, J. M. Rothberg, D. R. Link, N. Perrimon and M. L. Samuels, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14195–14200 CrossRef CAS PubMed.
- J. Kim, M. Hegde, S. H. Kim, T. K. Wood and A. Jayaraman, Lab Chip, 2012, 12, 1157–1163 RSC.
- J. Kim, H.-D. Park and S. Chung, Molecules, 2012, 17, 9818–9834 CrossRef CAS PubMed.
- T. Fujii, Microelectron. Eng., 2002, 61, 907–914 CrossRef.
- X. Niu, S. Peng, L. Liu, W. Wen and P. Sheng, Adv. Mater., 2007, 19, 2682–2686 CrossRef CAS PubMed.
- J. Park, T. Bansal, M. Pinelis and M. M. Maharbiz, Lab Chip, 2006, 6, 611–622 RSC.
- L. Wang, W. Liu, Y. Wang, J.-c. Wang, Q. Tu, R. Liu and J. Wang, Lab Chip, 2013, 13, 695–705 RSC.
- P. Abbyad, P.-L. Tharaux, J.-L. Martin, C. N. Baroud and A. Alexandrou, Lab Chip, 2010, 10, 2505–2512 RSC.
- N. L. Jeon, S. K. Dertinger, D. T. Chiu, I. S. Choi, A. D. Stroock and G. M. Whitesides, Langmuir, 2000, 16, 8311–8316 CrossRef CAS.
- M. Polinkovsky, E. Gutierrez, A. Levchenko and A. Groisman, Lab Chip, 2009, 9, 1073–1084 RSC.
- G. M. Walker, J. Sai, A. Richmond, M. Stremler, C. Y. Chung and J. P. Wikswo, Lab Chip, 2005, 5, 611–618 RSC.
- Y. S. Heo, L. M. Cabrera, J. W. Song, N. Futai, Y.-C. Tung, G. D. Smith and S. Takayama, Anal. Chem., 2007, 79, 1126–1134 CrossRef CAS PubMed.
- A. L. Paguirigan and D. J. Beebe, BioEssays, 2008, 30, 811–821 CrossRef CAS PubMed.
- J. W. Song, S. P. Cavnar, A. C. Walker, K. E. Luker, M. Gupta, Y.-C. Tung, G. D. Luker and S. Takayama, PLoS One, 2009, 4, e5756 Search PubMed.
- E. W. Young and D. J. Beebe, Chem. Soc. Rev., 2010, 39, 1036–1048 RSC.
- G. Walker, M. Ozers and D. Beebe, Biomed. Microdevices, 2002, 4, 161–166 CrossRef CAS.
- J. Khandurina, T. E. McKnight, S. C. Jacobson, L. C. Waters, R. S. Foote and J. M. Ramsey, Anal. Chem., 2000, 72, 2995–3000 CrossRef CAS.
- D. Schäpper, S. M. Stocks, N. Szita, A. E. Lantz and K. V. Gernaey, Chem. Eng. J., 2010, 160, 891–898 CrossRef PubMed.
- J. P. Urbanski, W. Thies, C. Rhodes, S. Amarasinghe and T. Thorsen, Lab Chip, 2006, 6, 96–104 RSC.
- R. H. Lam, M.-C. Kim and T. Thorsen, Anal. Chem., 2009, 81, 5918–5924 CrossRef CAS PubMed.
- R. J. Fish and E. K. Kruithof, BMC Mol. Biol., 2004, 5, 9 CrossRef PubMed.
- S. Gillis and K. A. Smith, Nature, 1977, 268, 154–156 CrossRef CAS.
- B. D. Walker, C. Flexner, K. Birch-Limberger, L. Fisher, T. J. Paradis, A. Aldovini, R. Young, B. Moss and R. T. Schooley, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9514–9518 CrossRef CAS.
- G. Mauleon, C. P. Fall and D. T. Eddington, PLoS One, 2012, 7, e43309 CAS.
- K. Funamoto, I. K. Zervantonakis, Y. Liu, C. J. Ochs, C. Kim and R. D. Kamm, Lab Chip, 2012, 12, 4855–4863 RSC.
- E. Leclerc, Y. Sakai and T. Fujii, Biotechnol. Prog., 2004, 20, 750–755 CrossRef CAS PubMed.
- M. Higuchi, J. Gen. Microbiol., 1984, 130, 1819–1826 CAS.
- M. Martin, R. Strachan, H. Aranha, S. Evans, M. Salin, B. Welch, J. Arceneaux and B. Byers, J. Bacteriol., 1984, 159, 745–749 CAS.
- S.-J. Ahn, Z. T. Wen and R. A. Burne, J. Bacteriol., 2007, 189, 8519–8527 CrossRef CAS PubMed.
- R. P. Ellen, Infect. Immun., 1976, 14, 1119–1124 CAS.
- J. Cisar, P. Kolenbrander and F. McIntire, Infect. Immun., 1979, 24, 742–752 CAS.
- D. J. Bradshaw, P. D. Marsh, G. K. Watson and C. Allison, Infect. Immun., 1998, 66, 4729–4732 CAS.
- J. Merritt, G. Niu, T. Okinaga and F. Qi, Appl. Environ. Microbiol., 2009, 75, 7725–7733 CrossRef CAS PubMed.
- R. Huang, M. Li and R. L. Gregory, Virulence, 2011, 2, 435–444 CrossRef PubMed.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 CrossRef CAS PubMed.
- E. L. Thomas and K. A. Pera, J. Bacteriol., 1983, 154, 1236–1244 CAS.
- M. Ibrahim and H. G. Schlegel, Biotechnol. Bioeng., 1980, 22, 1877–1894 CrossRef CAS PubMed.
- M. De Jong, J. Van der Hoeven, C. Van den Kieboom and P. Camp, FEMS Microbiol. Lett., 1988, 53, 45–51 CrossRef CAS.
- D. De Beer, P. Stoodley, F. Roe and Z. Lewandowski, Biotechnol. Bioeng., 1994, 43, 1131–1138 CrossRef CAS PubMed.
- M. B. Miller and B. L. Bassler, Annu. Rev. Microbiol., 2001, 55, 165–199 CrossRef CAS PubMed.
- S. Neethirajan, D. Karig, A. Kumar, P. P. Mukherjee, S. T. Retterer and M. J. Doktycz, in Encyclopedia of Nanotechnology, Springer, 2012, pp. 213–219 Search PubMed.
- S. Voloshin and A. Kaprelyants, Biochemistry (Moscow), 2004, 69, 1268–1275 CrossRef CAS PubMed.
- R. S. Zitomer and C. V. Lowry, Microbiol. Rev., 1992, 56, 1–11 CAS.
- T. P. Dalton, H. G. Shertzer and A. Puga, Annu. Rev. Pharmacol. Toxicol., 1999, 39, 67–101 CrossRef CAS PubMed.
- R. H. Wenger, J. Exp. Biol., 2000, 203, 1253–1263 CAS.
- J. Haaber, M. T. Cohn, D. Frees, T. J. Andersen and H. Ingmer, PLoS One, 2012, 7, e41075 CAS.
- R. Lamont and B. Rosan, Infect. Immun., 1990, 58, 1738–1743 CAS.
- J. Cisar, S. Curl, P. Kolenbrander and A. Vatter, Infect. Immun., 1983, 40, 759–765 CAS.
- A. L. Sandberg, S. Ruhl, R. A. Joralmon, M. J. Brennan, M. J. Sutphin and J. O. Cisar, Infect. Immun., 1995, 63, 2625–2631 CAS.
- P. E. Kolenbrander, Annu. Rev. Microbiol., 2000, 54, 413–437 CrossRef CAS PubMed.
- G. O'Toole, H. B. Kaplan and R. Kolter, Annu. Rev. Microbiol., 2000, 54, 49–79 CrossRef CAS PubMed.
- A. Tahmourespour, R. K. Kermanshahi, R. Salehi and N. Ghasemipero, Afr. J. Microbiol. Res., 2010, 4, 1051–1056 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra01577k |
| This journal is © The Royal Society of Chemistry 2014 |