
Digitally-patterned nanoprobe arrays for single cell insertion enabled by wet tapping
Abstract
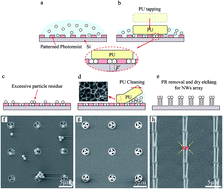
Wet tapping of nanoparticles for precisely-patterned nanoprobe arrays was developed to fabricate cell insertion tools for parallel multiple cell probing. Simple and scalable patterning of nanoparticles with patterning yield higher than 98% on a photoresist template was achieved by wet tapping. The effect of tapping time, particle materials, particle concentration, and surface conditions on the wet tapping was analyzed. Using the patterned nanoparticles, arrays of precisely-patterned nanoprobes were created. For cellular probing at single cell resolution, insertion of a controlled number of nanoprobes into single algal cells in a parallel fashion was also demonstrated.
 Please wait while we load your content...
Please wait while we load your content...

