
Synthesis and characterization of a novel organophosphorus oligomer and its application in improving flame retardancy of epoxy resin
Abstract
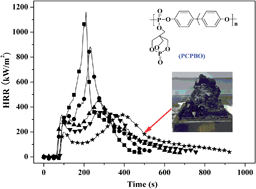
A novel organophosphorus, poly(4,4-dihydroxy-1-methyl-ethyl diphenol-o-bicyclic pentaerythritol phosphatephosphate) (PCPBO), was synthesized and characterized by FTIR, 1H NMR and 31P NMR. The flame retardancy and thermal stability of epoxy resin with different PCPBO loadings were investigated using the limited oxygen index (LOI), vertical burning test, cone calorimeter test and thermogravimetric analysis. The results showed that the incorporation of PCPBO into epoxy resin (EP) significantly improved its flame retardancy and thermal stability. The reduction of the peak heat release rate, total heat release and the increased char yield at high temperature further confirmed the improvement of the flame retardancy. FT-IR at different temperatures and the scanning electron microscopy of residual char revealed that the addition of PCPBO could induce the formation of an intumescent char layer, which retarded the degradation and combustion process of EP.
 Please wait while we load your content...
Please wait while we load your content...

