
Biodegradable multiblock polyurethane micelles with tunable reduction-sensitivity for on-demand intracellular drug delivery†
Abstract
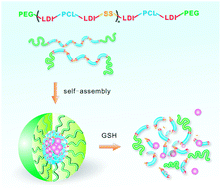
Redox-responsive nanovehicles containing disulfide bonds are particularly promising for targeted intracellular drug delivery. However, conventional reduction-sensitive nanocarriers generally lack control of stimuli-responsiveness due to their poor structural tunability. In this study, we developed a class of biodegradable multiblock polyurethanes bearing varied amounts of disulfide linkages in their backbone. The reducible polyurethanes exhibit interesting phase behavior and self-assembly properties, as well as triggered release profiles under an intracellular reduction-mimicking environment. It was found that the redox-sensitive polyurethane micelles can rapidly enter tumor cells and efficiently transport the encapsulated payloads into the cytosol. In vitro cytotoxicity studies demonstrated that the paclitaxel-loaded polyurethane micelles could inhibit the proliferation of tumor cells effectively, with the inhibition effects controlled by adjusting the disulfide content in the polymeric backbone. In addition, the drug-free nanomicelles possess good cytocompatibility toward both cancer cells and healthy cells. These multiblock bioresponsive polyurethanes hold great promise in the further development of controllable intracellular drug transporters.
 Please wait while we load your content...
Please wait while we load your content...

