
Flash carboxylation: fast lithiation–carboxylation sequence at room temperature in continuous flow†
Abstract
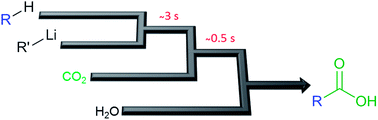
A method for the direct lithiation of terminal alkynes and heterocycles with subsequent carboxylation in a continuous flow format was developed. This method provides carboxylic acids at ambient conditions within less than five seconds with only little excess of the organometallic base and CO2.
 Please wait while we load your content...
Please wait while we load your content...

