
IM-17: a new zeolitic material, synthesis and structure elucidation from electron diffraction ADT data and Rietveld analysis†
Abstract
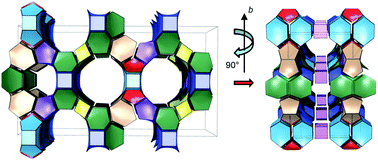
The synthesis and the structure of IM-17, a new germanosilicate with a novel zeolitic topology, prepared hydrothermally with decamethonium as the organic structure directing agent, are reported. The structure of calcined and partially rehydrated IM-17 of chemical formula per unit cell |(H2O)14.4|[Si136.50Ge39.50O352] was solved ab initio using electron diffraction ADT data in the acentric Amm2 (setting Cm2m) space group and refined by the Rietveld method. This new zeolite framework type contains a 3D pore system made of intersecting 12, 10 and 8-ring channels.
 Please wait while we load your content...
Please wait while we load your content...

