
A new route to dithia- and thiaoxacyclooctynes via Nicholas reaction†
Abstract
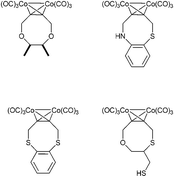
The current paper describes a new synthesis of heteroatom-substituted cyclooctynes. By using the Nicholas reaction we managed to design a concise synthesis that only uses three steps to build the eight-membered ring. It was also possible to functionalize said alkyne with a fluorophore.
 Please wait while we load your content...
Please wait while we load your content...

