
The synthesis of cyclohexenone using l-proline immobilized on a silica gel catalyst by a continuous-flow approach†
Abstract
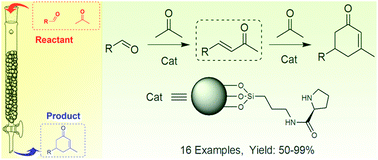
A facile and convenient method for the synthesis of cyclohexenone compounds was developed using an L-proline immobilized silica gel catalyst combined with a continuous-flow approach. Because of the mild reaction conditions, ease of catalyst recyclability, and product isolation, this reaction approach can potentially be used in a facile scale-up reaction or in industrial applications.
 Please wait while we load your content...
Please wait while we load your content...

