Interaction of single-stranded DNA with graphene oxide: fluorescence study and its application for S1 nuclease detection†
Yue He*ab,
Bining Jiao*ab and
Hongwu Tangc
aLaboratory of Quality & Safety Risk Assessment for Citrus Products (Chongqing), Ministry of Agriculture, Citrus Research Institute, Southwest University, Chongqing, 400712, China. E-mail: yuehe@cric.cn; jiaobining@cric.cn; Fax: +86 23 68349046; Tel: +86 23 68349046
bNational Citrus Engineering Research Center, Chongqing, 400712, China
cKey Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Sciences, Research Center for Nanobiology and Nanomedicine (MOE 985 Innovative Platform), Wuhan University, Wuhan, 430072, China. E-mail: hwtang@whu.edu.cn; Fax: +86 27 68754685; Tel: +86 27 68756759
First published on 8th April 2014
Abstract
As a new, water-soluble material, graphene oxide (GO) has gained growing interest for sensing applications. Particularly interesting is the interaction of nucleic acids with GO. Recently, it was found that short single-stranded DNA (ssDNA) had weaker affinity to GO than long ssDNA. This property makes it possible to prepare a novel bioassay platform for metal ions, antibiotics, and nuclease detection via the DNA(RNA) cleavage reaction. While practical analytical applications have been successfully demonstrated, few studies are focused on the mechanism of this phenomenon. In this work, we use fluorescence spectroscopy to deeply investigate the binding mechanism of ssDNA with GO to reveal the reason for this affinity difference caused by DNA length. Through computing with literature models, the main binding force, the binding constant, and number of binding sites between ssDNA and GO are obtained. Besides, our results show that the binding constant of short ssDNA with GO is much lower than that of long ssDNA with GO, which is the strongest evidence to prove the affinity difference between short ssDNA and long ssDNA with GO. Finally, based on these basic understandings of the interaction between ssDNA and GO, we develop a GO based biosensor for S1 nuclease and an inhibitor of S1 nuclease with satisfying results.
Introduction
Nanomaterials possess unique optical, electronic, magnetic, catalytic, mechanical, and thermal properties, which make them ideal candidates for signal generation and transduction in developing novel sensing systems with advanced and powerful functions. Graphene, as a kind of promising single-atom thick and two-dimensional nanomaterial, has received much attention.1,2 Particularly, graphene oxide (GO), which is a water-soluble derivative of graphene, has attracted increasing interest in making DNA-based optical sensors because of its unique characteristics such as good water dispersibility, facile surface modification, and high mechanical strength.3 Besides, GO can nonconvalently interact with single-stranded DNA (ssDNA) by π–π stacking interactions between nucleotide bases and GO.4 Furthermore, GO was reported to be a fluorescence superquencher with the long-range nanoscale energy transfer property,5,6 which, in combination with the unique GO–DNA interactions, has been employed to develop sensing systems for the detection of numerous important molecules.7–9 These fluorescence assays based on GO have shown great advantages. However, most of such sensing systems are based on the fact that the ssDNA and its rigid duplex or aptamer–target complexes exhibit different affinity to GO.Recently, Zhao et al. for the first time discovered that short ssDNA had weaker affinity to GO than long ssDNA.10 Based on this remarkable affinity difference, the GO–DNA complex has emerged as a novel bioassay platform to kick-start ultra-high sensitive mental ions,11 antibiotics,12 and nucleases13 detection via DNA(RNA) cleavage reaction, which greatly increasing the sensing application of GO. These design strategies are extremely simple: a FAM-labeled DNAzyme–substrate hybrid or a FAM-labeled long ssDNA substrate acted as both a molecular recognition module and signal reporter and GO as a superquencher. By taking advantage of the remarkable difference in affinity of GO with ssDNA containing a different number of bases in length, these proposed biosensors exhibits a high sensitivity towards the targets, which are much lower than previously reported optical biosensors.
While practical analytical applications of GO in the sensing system based on DNA(RNA) cleavage reaction have been successfully demonstrated, few studies are focused on the mechanism of the remarkable difference in affinity of GO with ssDNA containing a different number of bases in length. At the very start, research work has focused on measuring the force required to peel ssDNA molecules from single-crystal graphite using chemical force microscopy. It was found out that polythymine bind more strongly than polycytosine.14 After that, more work has focused on characterization of the adsorption of single nucleotides or nucleosides by atomic force microscopy (AFM),15,16 isothermal titration calorimetry,17 and theoretical calculations.18 However, none of these work investigated the effects of the length of ssDNA on the binding affinity between ssDNA and GO. It is particularly gratifying that Wu et al. started this work, they compared the adsorption of 12-, 18-, 24-, and 36-mer ssDNA on GO, and noticed that the quenching efficiency was lower for the longer ssDNA, suggesting weaker binding.19 It seems that Wu et al. and Zhao et al. have totally opposite conclusions to this problem.10,19 The reason may be that they compared the affinity in different DNA length regions, for Zhao et al. disposed towards shorter DNA length, even DNA fragments. Besides, in these studies,10–13,19 they only proved the affinity difference by fluorescence quenching ratio of GO to ssDNA with different lengths, the mechanism for this phenomenon still did not been systematically investigated. We believe such studies can serve as a basis for further design and optimization of GO and DNA(RNA) cleavage reaction-based biosensors. In this study, we'll provide complementary information to understand the effects of the length of ssDNA on the binding affinity between ssDNA and GO. In addition, based on above work, we'll design a biosensor for S1 nuclease detection to support our conclusion.
Experimental section
Chemicals and materials
The FAM-labeled 20-mer ssDNA with a sequence of 5′-FAM-TATATGGATGATGTGGTATT-3′ (20F), FAM-labeled 10-mer ssDNA with a sequence of 5′-FAM-TATATGGATG-3′ (10F), and FAM-labeled 5-mer ssDNA with a sequence of 5′-FAM-TATAT-3′ (5F), were synthesized by Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China). Graphene oxide was purchased from Sinocarbon Materials Technology Co., Ltd. (China). S1 nuclease, exonuclease I (Exo I), micrococcal nuclease (MNase), deoxyribonuclease I (DNase I) and exonuclease III (Exo III) were purchased from Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China). The buffer solutions used in this work are as follows: S1 nuclease buffer consisted of 40 mM CH3COONa–CH3COOH (pH 4.5), 300 mM NaCl, and 2 mM ZnCl2, and the Tris–HCl buffer consisted of 20 mM Tris–HCl (pH 7.4), 100 mM NaCl, 5 mM KCl and 5 mM MgCl2. Milli-Q purified water was used to prepare all the solutions.Apparatus
Fluorescent emission spectra were performed on Varian cary eclipse fluorescence spectrophotometer, Varian Medical Systems, Inc. (Palo Alto, American). The sample cell is a 700 μL quartz cuvette. The luminescence intensity was monitored by exciting the sample at 480 nm and measuring the emission at 520 nm. The slits for excitation and emission were set at 5 nm, 10 nm respectively. The fitting of the experimental data was accomplished using the software Origin 8.0.Interaction of single-stranded DNA (ssDNA) with graphene oxide (GO)
Fluorescence measurements were carried out by keeping the concentration of ssDNA fixed at 40 nM and that of GO was varied from 0.25 to 1.5 μg mL−1. Fluorescence spectra were recorded at 288, 298 and 308 K in the range of 500–640 nm upon excitation at 480 nm in each case (n = 3 replicates).Performance of S1 nuclease detection
For S1 nuclease assays, 2 μL of the ssDNA stock solution (10 μM), and appropriate concentrations of S1 nuclease solution were mixed, the mixed solution was diluted with CH3COONa–CH3COOH buffer (pH 4.5) to 20 μL. The above prepared solution was incubated for 30 min at 37 °C. Then 30 μL GO solution (100 μg mL−1) as prepared was added to the solution, the mixed solution was diluted with Tris–HCl (pH 7.4) buffer to 500 μL. The above prepared solution was incubated for 10 min at room temperature. Finally, the fluorescence intensity of the incubated solution was measured at 520 nm with excitation at 480 nm. For each concentration of the nuclease, the measurement has been repeated for at least three times independently. The error was calculated by the standard deviation of each concentration of the nuclease by the following formula:where n is the number of measurement of each concentration of the nuclease, xi is the measured values of each concentration of the nuclease,
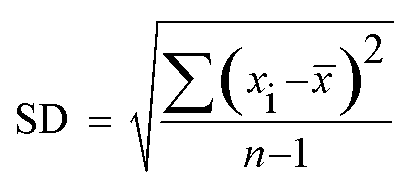
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) is the mean of the measured values of each concentration of the nuclease, and SD is the standard deviation of each concentration of the nuclease.
is the mean of the measured values of each concentration of the nuclease, and SD is the standard deviation of each concentration of the nuclease.
Results and discussion
Fluorescence quenching study
In this study, we employed three FAM-labeled single-stranded DNA (ssDNA) with DNA lengths of 20-, 10- and 5-mer, (named 20F, 10F and 5F, respectively) to systematically investigate the mechanism of the effect of ssDNA length on the binding affinity between ssDNA and graphene oxide (GO). None of these sequences can form highly stable secondary structures under experimental conditions, and the difference in performance is therefore expected to be caused by their length. All the FAM labels are on the 5′-end of the ssDNAs.To study the effect of DNA length on the binding affinity between ssDNA and GO, the binding characteristics of ssDNA with GO such as the quenching mechanism, the main binding force, the binding constant, and number of binding sites should be fully understood. We firstly study the effect of GO on fluorescence quenching of 20F. Fluorescence spectra of 20F in the absence, and presence of GO in Tris–HCl buffer were measured, respectively. With the addition of GO, a remarkable fluorescence decrease was observed. The fluorescence of 20F was quenched up to 96% of its original signal in the presence of 6 μg mL−1 GO (Fig. 1A). This is consistent with previous reports that GO can effectively quench the adsorbed FAM-ssDNA emission.20–24
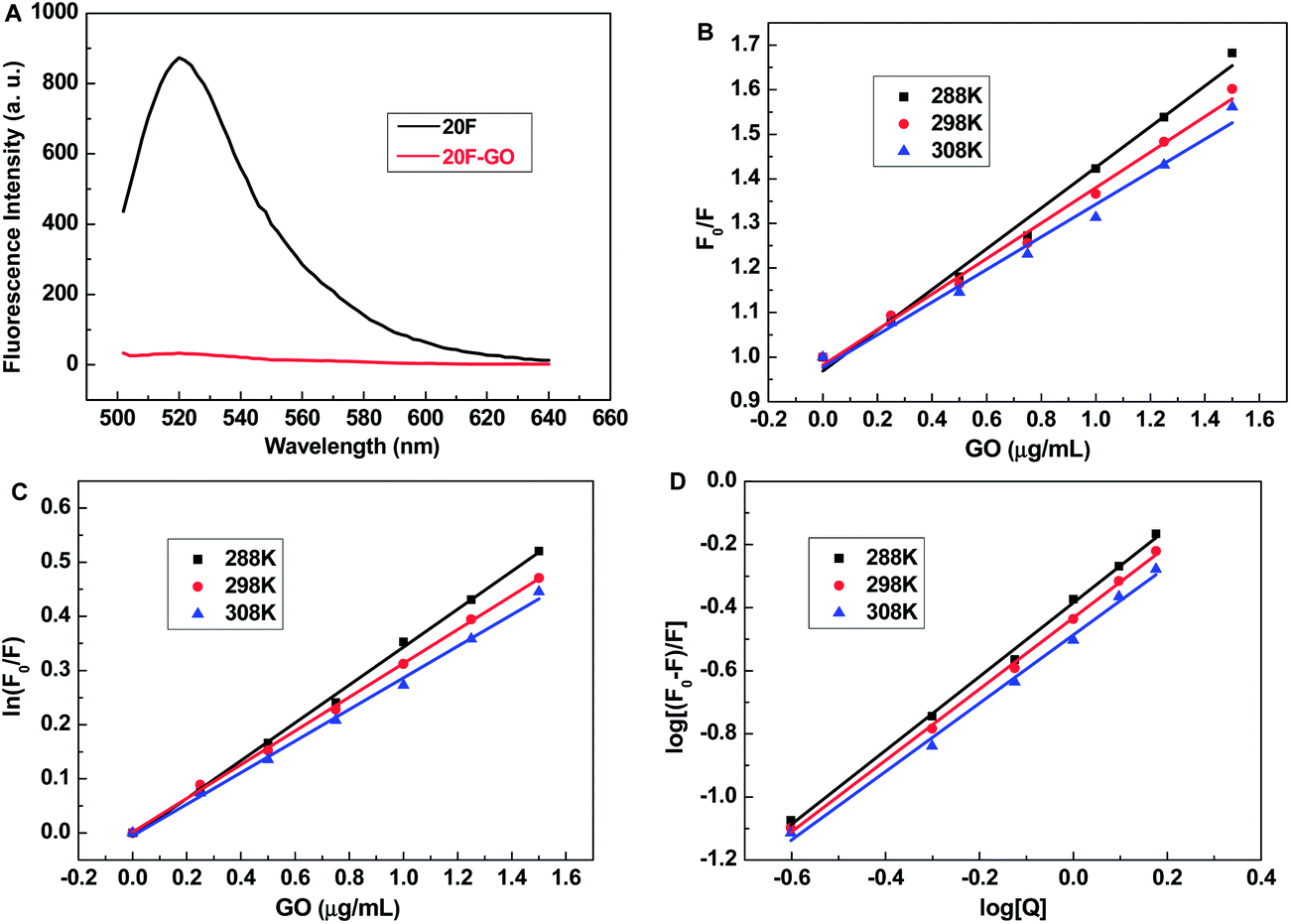 | ||
| Fig. 1 (A) Fluorescence spectra of 20F and 20F–GO in 20 mM Tris–HCl buffer (pH 7.4, 100 mM NaCl, 5 mM KCl, 5 mM MgCl2). [20F] = 40 nM, [GO] = 6 μg mL−1, λex = 480 nm. (B) Stern–Volmer plot for the binding of 20F with GO at 288 K, 298 K and 308 K; (C) Perrin plot for the binding of 20F with GO at 288 K, 298 K and 308 K; (D) plots of log[(F0 − F)/F] vs. log[Q] for the binding of 20F with GO at 288 K, 298 K and 308 K. | ||
To quantitatively analyze the quenching of 20F by GO, we use fluorescence spectroscopy to study the quenching mechanism between 20F and GO. As we all know, there are two quenching processes: static and dynamic quenching.25 Dynamic quenching results from the diffusive encounter between quencher and fluorophore during the lifetime of the excited state; static quenching results from the formation of a non-fluorescent ground-state complex (fluorophore–quencher). Dynamic and static quenching can be distinguished based on their differences on temperature dependence. Higher temperature results in faster diffusion and larger amounts of collisional quenching. It will typically lead to the dissociation of weakly bound complexes and smaller amounts of static quenching. Therefore, the quenching constant increases for dynamic quenching while it decreases for static quenching with increase in temperature. The equation for dynamic quenching is presented by (1):
| F0/F = 1 + KSV[Q] | (1) |
In this equation, F0 and F are the emission intensities of 20F in the absence and presence of GO, respectively; KSV is the Stern–Volmer constant, which characterizes the dynamic quenching efficiency of the quencher; and [Q] is the concentration of the quencher. The change in F0/F of 20F with GO concentration is shown in Fig. 1B. At the low GO concentration range (0–1.5 μg mL−1), the Stern–Volmer plots were observed to be linear for 20F–GO with the slopes decreasing with increase in temperatures. The values of KSV and R at different temperatures were evaluated which are given in Table 1. The values of KSV at different temperatures indicate the presence of static quenching mechanism in the interaction between 20F and GO.
| ssDNA | Temperature (K) | KSV (R)b (mL μg−1) | KP (R)b (mL μg−1) | KA (R)b (mL μg−1) | n | ΔH | ΔG | ΔS |
|---|---|---|---|---|---|---|---|---|
| a Each sample was analyzed in triplicate, and the results are the average values.b R is the correlation coefficient. | ||||||||
| 20F | 288 | 0.4568 (0.9902) | 0.3496 (0.9983) | 0.4123 (0.9969) | 1.17 | <0 | <0 | >0 |
| 298 | 0.3982 (0.9927) | 0.3119 (0.9989) | 0.3708 (0.9912) | 1.05 | <0 | |||
| 308 | 0.3659 (0.9859) | 0.2919 (0.9967) | 0.3150 (0.9926) | 1.10 | <0 | |||
| 10F | 288 | 0.2821 (0.9959) | 0.2363 (0.9983) | 0.2630 (0.9985) | 1.18 | <0 | <0 | >0 |
| 298 | 0.2593 (0.9928) | 0.2202 (0.9974) | 0.2354 (0.9984) | 1.19 | <0 | |||
| 308 | 0.2193 (0.9960) | 0.1898 (0.9994) | 0.2071 (0.9993) | 1.09 | <0 | |||
| 5F | 288 | 0.1333 (0.9933) | 0.1215 (0.9968) | 0.1254 (0.9982) | 1.08 | <0 | <0 | >0 |
| 298 | 0.0923 (0.9994) | 0.0864 (0.9993) | 0.0923 (0.9994) | 0.99 | <0 | |||
| 308 | 0.0561 (0.9992) | 0.0539 (0.9991) | 0.0552 (0.9992) | 1.05 | <0 | |||
Next, in order to invoke the possibility of the presence of static quenching mechanism in the interaction between 20F and GO, we calculated the static quenching constant by the eqn (2) for static quenching:
| ln(F0/F) = KP[Q] | (2) |
In this equation, KP is the Perrin constant, which characterizes the static quenching efficiency of the quencher. The change in ln(F0/F) of 20F with GO concentration is shown in Fig. 1C. At the low GO concentration range (0–1.5 μg mL−1), the Perrin plots was observed to be linear for 20F–GO with the slopes decreasing with increase in temperatures. The values of KP and R at different temperatures were evaluated which are given in Table 1. This result supports our argument that the quenching was not initiated by dynamic collision but originated from the formation of a complex.
Binding constants and binding sites
For the static quenching, the binding constant KA and the number of binding sites n could be represented by the equation:26
 | (3) |
The change in log[(F0 − F)/F] of 20F with log[Q] is shown in Fig. 1D. At the low GO concentration range (0–1.5 μg mL−1), the values of log[(F0 − F)/F] were observed to be linear for the values of log[Q] with the slopes decreasing with increase in temperatures. The values of KA and n at different temperatures for 20F–GO were calculated from the intercept and slope of the plots of log[(F0 − F)/F] versus log[Q], which are listed in Table 1. The KA values decreased with the increasing temperature implied the complex of 20F–GO became less stable at higher temperature, which further evidenced that the fluorescence quenching was a static quenching process.
It is noteworthy that K data are usually expressed in L mol−1 units, however, in our work, we presented them in mL μg−1. The reason is that the accurate molecular weight of the GO can't be determined for the structural heterogeneities of GO. Then, the question is: does this units influence the analysis. In fact, the units form of K data is not so important in our work, the changing trend of K data with increase of temperature is what our concern.
Thermodynamic parameters and nature of the binding forces
The thermodynamic parameters, ΔH, ΔG and ΔS of ssDNA–GO interaction are important for confirming binding mode, where the values of ΔH, ΔG and ΔS are enthalpy change, free energy change and entropy change. In general, acting forces between small molecular and biomacromolecule mainly include hydrogen bonds, van der Waals forces, electrostatic interactions, hydrophobic forces, etc. By the values of the binding constants KA at 288, 298 and 308 K, the thermodynamic parameters such as ΔH, ΔG and ΔS could be estimated according to the following equations:
 | (4) |
ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKA lnKA
| (5) |
 | (6) |
However, the accurate molecular weight of the GO can not be determined, we only can determine a formula weight (45.1 g mol−1) from the empirical formula of GO (C2.01H1.00O1.25) (shown in ESI, Table S1†). But the formula weight for the GO is not necessarily the molecular weight. As we all know, a molecular formula is the same as or a multiple of the empirical formula, and is based on the actual number of atoms of each type in the compound. For example, if the empirical formula of a compound is C3H8, its molecular formula may be C3H8, C6H16, etc. So, we can conclude that the molecular weight of GO is the same as or a multiple of 45.1 g mol−1. Based on above analysis, we can roughly determine the sign of ΔH, ΔG and ΔS from eqn (4)–(6). As the KA values decreased with the increasing temperature, we can conclude ΔH is negative from the eqn (4); as the molecular weight of the GO is the same as or a multiple of 45.1 g mol−1, we can compute KA from the eqn (3) which has a very big value, so ΔG is negative from eqn (5); as the ΔH and ΔG symbols are determined, we can conclude ΔS is positive from the eqn (6). The calculated thermodynamic parameters for the interaction between 20F and GO are listed in Table 1. The negative ΔG value means that the interaction process between 20F and GO was spontaneous. According to the point of view of Ross and Subramanian,27 when ΔH < 0 or ΔH ≈ 0, ΔS > 0, the main binding force was electrostatic force; when ΔH < 0, ΔS < 0, the main binding force was van der Waals force or hydrogen bond and when ΔH > 0, ΔS > 0, the main binding force was hydrophobic force. So the results indicated that electrostatic force was the main binding force to stabilize the complex of 20F–GO in Tris–HCl buffer (pH = 7.4, 100 mM NaCl, 5 mM KCl, 5 mM MgCl2).
This phenomenon may result from the structure of GO. The presence of ionic groups and aromatic domains suggests that GO can interact with ssDNA in a number of ways. Ionic groups such as O– and COO– that decorate the planes and edges of GO allow electrostatic interactions with ssDNA, and the aromatic scaffold provides a platform for π–π stacking and quenching of dyes. Especially in ionic buffer, metal ions act as a bridge to connect these two negatively charged molecules.
Effects of the length of single-stranded DNA (ssDNA)
According to the above method, we following study the effect of GO on fluorescence quenching of 10F and 5F, respectively.The change in F0/F of 10F and 5F with GO concentration are shown in Fig. S1A and S2A,† respectively. At the low GO concentration range (0–1.5 μg mL−1), the Stern–Volmer plots were observed to be linear for 10F–GO and 5F–GO with the slopes decreasing with increase in temperatures. The change in ln(F0/F) of 10F and 5F with GO concentration are shown in Fig. S1B and S2B,† respectively. The Perrin plots were observed to be linear for 10F–GO and 5F–GO with the slopes decreasing with increase in temperatures. The values of KSV, KP and R at different temperatures were evaluated which are given in Table 1. These results indicate the presence of static quenching mechanism in the interaction of 10F with GO and 5F with GO.
The change in log[(F0 − F)/F] of 10F and 5F with log[Q] are shown in Fig. S1C and S2C,† respectively. The values of log[(F0 − F)/F] were observed to be linear for the values of log[Q] with the slopes decreasing with increase in temperatures for both 10F and 5F. The values of KA and n at different temperatures for 10F–GO and 5F–GO were calculated from the intercept and slope of the plots of log[(F0 − F)/F] versus log[Q], respectively, which are listed in Table 1. The KA values decreased with the increasing temperature implied the complex of 10F–GO and 5F–GO became less stable at higher temperature, which further evidenced that the fluorescence quenching was a static quenching process. Meanwhile, we notice that the KA values between GO and ssDNA is strongly affected by the length of ssDNA: the KA value of short ssDNA with GO is much lower than that of long ssDNA with GO under the same temperature, which means the affinity of the short ssDNA to GO is significantly weaker than that of the long ssDNA.
We also calculated the thermodynamic parameters for the interaction of 10F and 5F with GO, respectively, which are listed in Table 1. The results indicated that electrostatic force was also the main binding force to stabilize the complex of 10F–GO and 5F–GO in Tris–HCl buffer.
The above results indicate that either short ssDNA or long ssDNA, the quenching mechanism is static quenching and the main binding force is electrostatic force. The only difference is the binding constant of short ssDNA with GO is much lower than that of long ssDNA with GO, which means the affinity of the short ssDNA to GO is significantly weaker than that of the long ssDNA, as reported by Zhao et al.10
S1 nuclease detection
On the basis of above analysis, we constructed a GO-based sensing system for endonuclease detection to support our conclusion. S1 nuclease, which exhibits endo- and exolytic hydrolytic activity for the phosphodiester bonds of ssDNA or RNA and produces mono- or oligonucleotide fragments,28,29 is taken as the model endonucleases to provide the “proof-of-principle” verification of this method. Fig. 2 illustrates the sensing strategy for the detection of S1 nuclease. In the absence of S1 nuclease, 20F which is used as the nuclease substrate is adsorbed onto the GO sheet by π–π stacking making the fluorophore close proximity to GO surface, thus GO significantly quenches the fluorescence of FAM. In the presence of S1 nuclease, the 20F is cut into fragments by S1 nuclease, the introduction of GO into the sensing solution results in weak quenching of the fluorescence of the FAM due to the weak affinity of the short FAM-linked oligonucleotide fragment to GO, and the fluorescence intensity gradually increases with increasing concentration of S1 nuclease. Therefore, the fluorescence intensity of FAM as a function of S1 nuclease concentration is measured correspondingly. | ||
| Fig. 2 Scheme for the mechanism of GO-based biosensor for S1 nuclease detection. | ||
To achieve the best sensing performance, the concentration of GO, the quenching reaction time between GO and 20F and S1 nuclease-catalyzed digestion reaction time, and the adding order of S1 nuclease and GO were optimized and the results were shown in ESI, Fig. S3, S4, S5,† respectively. The assay of S1 nuclease was carried out under the optimized conditions with the fixed concentrations of 20F (40 nM) and GO (6 μg mL−1). Fig. 3A shows the fluorescence emission spectra of the GO-based biosensor in the presence of different concentrations of S1 nuclease. The fluorescence intensity of the biosensor dramatically increases with the increasing concentration of S1 nuclease (shown in ESI, Table S2†). The calibration curve for S1 nuclease detection is shown in Fig. 3B, and the linear range is from 8.0 × 10−4–3.2 × 10−2 units mL−1 with linear equation y = 24715x + 21.66, where y is the fluorescence intensity of FAM at 520 nm and x is the concentration of S1 nuclease (regression coefficient R2 = 0.9936). The detection limit is estimated to be 5.8 × 10−4 units mL−1 (3S0/S, in which S0 is the standard deviation for the blank solution, n = 11, and S is the slope of the calibration curve), which is of much lower than those reported S1 nuclease optical biosensors (shown in ESI, Table S3†).30–35 A series of eleven repetitive measurements of 2.0 × 10−2 units mL−1 S1 nuclease were used for estimating the precision, and the relative standard deviation (RSD) was 3.7%, showing good reproducibility of the proposed method. Besides, the specificity of the sensing system (shown in ESI, Fig. S6†) and the determination of an inhibitor of S1 nuclease (shown in ESI, Fig. S7 and S8†) had satisfying results. This excellent performance for S1 nuclease detection supports our conclusion that short ssDNA had weaker affinity to GO than long ssDNA. Since DNA(RNA) cleavage reaction involve numerous nucleases, the remarkable affinity difference of ssDNA with GO caused by DNA length provides a new general platform for sensitive detection of various targets and could find wide applications in molecular diagnostics, genomic research, and drug development fields.
 | ||
| Fig. 3 Fluorescence emission spectra of GO-based biosensor in the presence of increasing amount of S1 nuclease and calibration curve for S1 nuclease detection. (A) Fluorescence emission spectra of GO-based biosensor in the presence of increasing amount of S1 nuclease. (B) Calibration curve for S1 nuclease detection. Excitation: 480 nm. | ||
Conclusions
In summary, we have systematically studied the interaction of single-stranded DNA (ssDNA) with graphene oxide (GO) by fluorescence spectroscopy. Stern–Volmer constant, Perrin constant, binding constant, thermodynamic parameters were computed using literature models. The results show that the quenching mechanism is static quenching, the main binding force is electrostatic force between GO and ssDNA, and the binding constant between GO and ssDNA is strongly affected by the length of ssDNA: the binding constant of short ssDNA with GO is much lower than that of long ssDNA with GO, which means the affinity of the short ssDNA to GO is significantly weaker than that of the long ssDNA. Finally, based on these basic understandings of the interaction between GO and ssDNA, a simple and ultra-high sensitive strategy for S1 nuclease detection using GO-based biosensor is developed. This GO-based biosensor is extraordinarily sensitive to S1 nuclease detection. As to S1 nuclease, a sensitive detection limit of 5.8 × 10−4 units mL−1 was obtained. We expect that this assay platform will become an important assay tool in drug screening and basic research related to endonucleases (ENases).Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (no. SWU113099) and the National Key Technology R&D Program (no. 2009BADB7B04) and China Agriculture Research System (no. CARS-27).Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- C. H. Lu, H. H. Yang, C. L. Zhu, X. Chen and G. N. Chen, Angew. Chem., Int. Ed. Engl., 2009, 48, 4785 CrossRef CAS PubMed.
- R. S. Swathi and K. L. Sebastian, J. Chem. Phys., 2008, 129, 054703 CrossRef CAS PubMed.
- R. S. Swathi and K. L. Sebastian, J. Chem. Phys., 2009, 130, 086101 CrossRef CAS PubMed.
- J. Lee, Y. K. Kim and D. H. Min, Anal. Chem., 2011, 83, 8906 CrossRef CAS PubMed.
- C. H. Lu, J. Li, X. J. Qi, X. R. Song, H. H. Yang, X. Chen and G. N. Chen, J. Mater. Chem., 2011, 21, 10915 RSC.
- Z. Zhou, C. Zhu, J. Ren and S. Dong, Anal. Chim. Acta, 2012, 740, 88 CrossRef CAS PubMed.
- X. H. Zhao, R. M. Kong, X. B. Zhang, H. M. Meng, W. N. Liu, W. H. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2011, 83, 5062 CrossRef CAS PubMed.
- M. Liu, H. M. Zhao, S. Chen, H. T. Yu, Y. B. Zhang and X. A. Quan, Biosens. Bioelectron., 2011, 26, 4111 CrossRef CAS PubMed.
- F. Li, Y. Feng, C. Zhao, P. Li and B. Tang, Chem. Commun., 2012, 48, 127 RSC.
- Y. He, L. H. Xiong, X. J. Xing, H. W. Tang and D. W. Pang, Biosens. Bioelectron., 2013, 42, 467 CrossRef CAS PubMed.
- S. Manohar, A. R. Mantz, K. E. Bancroft, C. Y. Hui, A. Jagota and D. V. Vezenov, Nano Lett., 2008, 8, 4365 CrossRef CAS.
- G. Wei, Q. Li, S. Steckbeck and L. C. Ciacchi, Phys. Chem. Chem. Phys., 2014, 16, 3995 RSC.
- B. S. Husale, S. Sahoo, A. Radenovic, F. Traversi, P. Annibale and A. Kis, Langmuir, 2010, 26, 18078 CrossRef PubMed.
- N. Varghese, U. Mogera, A. Govindaraj, A. Das, P. K. Maiti, A. K. Sood and C. N. Rao, ChemPhysChem, 2009, 10, 206 CrossRef CAS PubMed.
- J. Antony and S. Grimme, Phys. Chem. Chem. Phys., 2008, 10, 2722 RSC.
- M. Wu, R. Kempaiah, P. J. Huang, V. Maheshwari and J. W. Liu, Langmuir, 2011, 27, 2731 CrossRef CAS PubMed.
- H. X. Chang, L. H. Tang, Y. Wang, J. H. Jiang and J. H. Li, Anal. Chem., 2010, 82, 2341 CrossRef CAS PubMed.
- H. F. Dong, W. C. Gao, F. Yan, H. X. Ji and H. X. Ju, Anal. Chem., 2010, 82, 5511 CrossRef CAS PubMed.
- Y. Wang, Z. H. Li, D. H. Hu, C. T. Lin, J. H. Li and Y. H. Lin, J. Am. Chem. Soc., 2010, 132, 9274 CrossRef CAS PubMed.
- Y. Q. Wen, F. F. Xing, S. J. He, S. P. Song, L. H. Wang, Y. T. Long, D. Li and C. H. Fan, Chem. Commun., 2010, 46, 2596 RSC.
- L. F. Sheng, J. T. Ren, Y. Q. Miao, J. H. Wang and E. K. Wang, Biosens. Bioelectron., 2011, 26, 3494 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 1999, p. 277 Search PubMed.
- S. Bi, L. Ding, Y. Tian, D. Song, X. Zhou, X. Liu and H. Zhang, J. Mol. Struct., 2004, 703, 37 CrossRef CAS PubMed.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096 CrossRef CAS.
- F. Harada and J. E. Dahlberg, Nucleic Acids Res., 1975, 2, 865 CrossRef CAS PubMed.
- R. C. Wiegand, G. N. Godson and C. M. Radding, J. Biol. Chem., 1975, 250, 8848 CAS.
- Z. X. Zhou, J. B. Zhu, L. B. Zhang, Y. Du, S. J. Dong and E. K. Wang, Anal. Chem., 2013, 85, 2431 CrossRef CAS PubMed.
- X. J. Yang, F. Pu, J. S. Ren and X. G. Qu, Chem. Commun., 2011, 47, 8133 RSC.
- F. Pu, D. Hu, J. S. Ren, S. Wang and X. G. Qu, Langmuir, 2010, 26, 4540 CrossRef CAS PubMed.
- Y. Zhang, Y. Y. Wang and B. Liu, Anal. Chem., 2009, 81, 3731 CrossRef CAS PubMed.
- M. Liu, H. M. Zhao, S. Chen, H. T. Yu and X. Quan, ACS Nano, 2012, 6, 3142 CrossRef CAS PubMed.
- R. Cao, B. X. Li, Y. F. Zhang and Z. N. Zhang, Chem. Commun., 2011, 47, 12301 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra01102c |
| This journal is © The Royal Society of Chemistry 2014 |