
Electrochemical and in situ ATR-SEIRAS investigations of methanol and CO electro-oxidation on PVP-free cubic and octahedral/tetrahedral Pt nanoparticles
Abstract
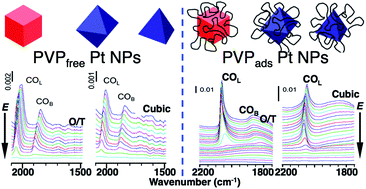
Most wet-chemical methods that synthesize metal nanoparticles (NPs) of a particular size and desired shape include the use of a stabilizing surfactant, such as poly(vinylpyrrolidone) (PVP). The latter has the ability to prohibit and/or promote surface sites from participating in electrocatalytic reactions, i.e. the methanol oxidation reaction (MOR). In light of our recent findings that adsorbed PVP can enhance the MOR on Pt NPs, a strong effort was made herein to separate the NP surface orientation effect from that of surface-bound PVP. We report the in situ ATR-SEIRAS (attenuated total reflection-surface enhanced infrared reflection absorption spectroscopy) and electrochemical (EC) studies of MOR and CO oxidation reaction (COR) performed on PVP-free cubic and octahedral/tetrahedral (O/T) Pt NPs that were obtained using an adapted liquid phase UV photo-oxidation (UVPO) technique. Transmission electron microscope (TEM) images showed no observable changes of shape and size after the elimination of the PVP, while the integrity of the atomic surface structure was further confirmed by the orientation-dependent EC stripping analysis of irreversibly adsorbed adatoms, i.e. Bi and Ge. The MOR activity was enhanced by the preferential surface orientation of the O/T Pt NPs compared to commercial Pt black and the cubic Pt NPs. The in situ ATR-SEIRAS measurements showed that the PVP-free Pt NPs adsorbed more bridge-bound gaseous CO than those with residual PVP and the weakly hydrogen-bound interfacial water played an important role for the orientation dependent enhancement in MOR activity. Additionally, the findings of this work are coupled with previously published investigations regarding the influence that PVP exerts on the ultimate activity of the Pt NPs, i.e., the adsorbed PVP enhances further the MOR activity on the O/T but suppresses it on the cubic Pt NPs.
 Please wait while we load your content...
Please wait while we load your content...

