
Simultaneously improving the tensile strength and modulus of aramid fiber by enhancing amorphous phase in supercritical carbon dioxide
Abstract
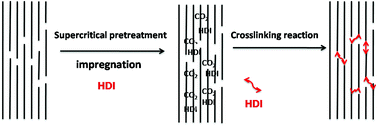
The aramid fiber made from poly(p-phenyleneterephthalamide) PPTA has high strength and high modulus due to a perfect orientation of rigid macromolecular chains. Many attempts to improve the mechanical performance of the aramid fiber were proposed during the last decade; however, simultaneously improving the strength and modulus of the aramid fiber is still a great challenge as most methods can only improve the modulus at the expense of reducing the tensile strength. The mechanism of broken aramid fibers indicates that the weak point of the aramid fiber is at amorphous phase. Therefore, if the amorphous phase can be enhanced, the mechanical performance of PPTA fibers would be improved. Here a novel method is reported to enhance the amorphous phase of PPTA fibers by treating the aramid fiber in supercritical carbon dioxide (ScCO2) with or without hexamethylene diisocyanate (HDI). In this new process, the ScCO2, with or without the crosslinker HDI in ScCO2, is diffused into the amorphous phase of PPTA fibers and the amorphous phase is enhanced by re-arrangement of molecular segments or HDI reacting with the groups (–NH2, –COOH, and –NHCO–) of PPTA fibers. This is done without breaking the crystalline structure and orientation by carefully controlling the reaction temperature, pressure and time. The results showed that the tensile strength and modulus had simultaneously increased. Molecular weight could be increased under appropriate treatment, while the crystalline structures of PPTA fibers were almost unchanged. Dynamic mechanical analysis (DMA) and thermomechanical analysis (TMA) showed that the glass-transition temperature had increased. All the results implied the chain crosslinking mainly took place in the amorphous phase of the PPTA fibers and the performance improvement was attributed to the amorphous enhancement.
 Please wait while we load your content...
Please wait while we load your content...

