DOI:
10.1039/C4RA00648H
(Paper)
RSC Adv., 2014,
4, 17860-17865
Continuous synthesis of triacetonamine over sulfonic acid-functionalized mesoporous silicas
Received
22nd January 2014
, Accepted 10th March 2014
First published on 3rd April 2014
Abstract
The continuous synthesis of triacetonamine from acetone and ammonia was realized over sulfonic acid-functionalized mesoporous silicas. These catalysts were characterized by TG, XPS, BET, elemental analysis and acid–base titration. The results indicated that both the acidity and the textural properties of the catalysts had significant influences on the catalytic performance. Meanwhile, the effects of reaction parameters on the reaction of acetone and ammonia were investigated. Based on the experimental results, the reaction mechanisms were speculated.
1. Introduction
Hindered amine light stabilizers (HALS) have been widely applied to polymeric materials for the exclusive effect of light stabilizers. Triacetonamine as the only origin of the 2,2,6,6-tetramethylpiperidine ring is the basic material of HALS and plays an important role in this field. As early as 1880, Heinz1 reported the synthesis of triacetonamine via the conversion of acetone to phorone and then reaction of phorone with ammonia. Then, Hall2 directly obtained triacetonamine from acetone and ammonia with CaCl2 in nine days. Obviously, the one-step process has distinct advantage over the two-step process. In order to improve the reaction efficiency, Ivan3–5 employed Lewis acids, protonic acids and their ammonium salts as catalysts for the preparation of triacetonamine from acetone and ammonia. Meanwhile, Tohru,6 Walter,7 Yutaka8 and Hartmut9 studied this reaction in the presence of acyl chloride, CCl4, TiCl4 and sulfonyl chloride. And even Me2SO4 and hydrazine hydrogen halide salts were evaluated as catalysts for the synthesis of triacetonamine by Manfred10 and Bunji,11 respectively. However, quite complicated reactions occurring in the system of acetone and ammonia make it very hard to enhance the selectivity of triacetonamine with the modest conversion of acetone.12 In addition, the aforementioned methods are all carried out in autoclave or flask. These tedious work-up procedures and contaminated operations restricted the development of triacetonamine. So it is very imperative to further select catalysts and change the traditional processes. Continuous synthesis of triacetonamine may be a good choice. However, there are few reports on this aspect.
In this paper, we try to establish a continuous process for the synthesis of triacetonamine from acetone and ammonia over sulfonic acid-functionalized mesoporous silicas in a fixed-bed reactor. These catalysts are characterized by thermogravimetric analysis (TG), X-ray photoelectron spectroscopy (XPS), element analysis, N2 adsorption and desorption experiments and acid–base titration to clarify their structure–activity relationships. Meanwhile, the influences of reaction parameters on the reaction of acetone and ammonia are investigated over functionalized catalysts. Based on the experimental results, the reaction mechanisms are speculated.
2. Experimental
2.1 Reagents
Tetraethoxysilane (TEOS, Tianjin Guangfu Fine Chemical Research Institute, China), 3-mecaptopropyltrimethoxysilane (MPTMS, Qufu Chenguang Chemical Co., Ltd, China), the poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) block copolymer templating agent Pluronic 123 (Shandong Deshi Chemical Co., Ltd, China), H2O2, concentrated hydrochloric acid and ethanol (Tianjin Jiangtian Chemical Co., Ltd, China) were all used as received.
2.2 Preparation of catalysts
The catalysts were prepared as follows: 20.0 g Pluronic 123 were dissolved with stirring in 490 g water at room temperature and then 8.2 mL concentrated hydrochloric acid was added to this solution under vigorous stirring. The solution was heated to 40 °C and 38.9 g TEOS were added. After prehydrolysis for 2 h, 7.0 g, 14.0 g, 21.0 g and 28.0 g MPTMS were respectively added to the above-mentioned reaction mixture followed by the addition of 42 mL 30 wt% H2O2. The reaction mixture was stirred for 20 h. The solid powder was obtained by filtration. Filtrate was treated with active carbon. The solid and 150 mL of filtrate were merged and aged at 90 °C in a polycarbonate bottle under static conditions for another 24 h. The product was recovered by filtration, refluxing in ethanol for 4 h thrice and then dried at 50 °C for 6 h. The dried solid powder was kneaded vigorously with 5 mL of acidic silica sol (25 wt%), followed by molding into a cylinder with diameters of 3–3.5 mm by use of an extruder. Then, the sample cylinders were dried at 65 °C for 12 h and cut into small pieces with lengths of about 5 mm. These obtained catalysts were denoted by S-1, S-2, S-3 and S-4, respectively.
2.3 Catalytic reaction
The condensation of acetone and ammonia was carried out in a tubular, fixed-bed reactor with an inner diameter of 15 mm and a length of 650 mm, which was charged with 40 mL catalysts. Acetone was dosed into the reactor by a syringe pump. The ammonia flow rate was set by an S49 33/MT mass gas flow controller. The temperature in the reaction zone was measured by a thermocouple located in the center of the tube and connected to a proportion integration differentiation cascade controller. The reaction mixture was analyzed by gas chromatography (GC) with a 30 m SE-54 capillary column and a Flame Ionization Detector (FID). Moreover, the components of the reaction mixture were identified by a gas chromatography-mass spectrometry (GCMS) equipped with an HP-5 capillary column (30 m × 0.25 mm, 0.2 μm film thickness) and an ion trap MS detector.
2.4 Catalyst characterization
Temperature–gravity properties of these catalysts, S-1, S-2 and S-3, were measured with an STA 409PC thermo gravimetric (TG) analyzer. The catalysts were heated from room temperature to 800 °C at a rate of 10 °C min−1 in a stream of N2 (40 mL min−1). X-ray photoelectron spectroscopy (XPS) was recorded on a PHI1600 spectrometer with an Mg Kα X-ray source for excitation. Acid–base titrations were used to measure acid capacity of the catalysts. Typically, 0.05 g catalyst was added to 10 mL 2 M NaCl solution and stirred overnight. This solution was then titrated with 0.01 M NaOH solution in the presence of phenolphthalein.13 Elemental analysis was carried out on a Vario Micro cube element analyzer. N2 adsorption and desorption experiments were performed in liquid nitrogen using a NOVA 2000e analyzer (Quantachrome, US). The total surface area (SBET) was calculated from the linear part of the Brunauer–Emmer–Teller (BET). The micro pore volume (Vmicro) and the external surface area (SEXT) were estimated by the t-plot. The pore-size distributions were obtained using the method of Barrett–Joyner–Halenda (BJH).
3. Results and discussion
3.1 Catalyst selection
As mentioned above, the reaction of acetone and ammonia is quite complicated. A number of reversible reactions proceed competitively at the same time. Many studies have demonstrated that several compounds, like triacetonamine, acetonine, phorone, diacetone alcohol, diacetonamine and mesityl oxide, are co-existed in the reaction mixture.14 Therefore, the low selectivity of triacetonamine has persecuted the manufacturers for a long time. In order to increase the selectivity of triacetonamine, the improvement of catalysts is crucial. Today, NH4NO3 is considered as a suitable catalyst for industrial production and its nature is a Brønsted acid. So a strong Brønsted acid may be efficient for the synthesis of triacetonamine. In addition, considering the excellent mechanical strength and stability of silicone, we tried to immobilize sulfonic acid on the silicone to yield catalysts S1, S2, S3 and S4. Their catalytic performances were investigated and compared with NH4NO3/γ-Al2O3, NH4NO3/SiO2, kinds of sulfonic acid resin and solid super acids, such as SO42−/ZrO2 and SO42−/TiO2. The NH4NO3 supported catalysts and the solid super acids exhibited shorter life due to the leaking of the active components. The sulfonic acid resins normally suffer from easy swelling, poor mechanical strength and low thermal stability, which inhibited the application in continuous synthesis of triacetonamine. However, the sulfonic acid-functionalized mesoporous silicas displayed excellent mechanical strength and thermal stability. Their catalytic performances were characterized and the obtained results were shown in Table 1. It was found that with the increase of MPTMS content, the acetone conversion increased from 28.7% to 44.7% and then decreased to 31.1%. The triacetonamine selectivity remained essentially unchanged. Catalyst S-2 exhibited better catalytic performance than others. So it was speculated that not just the acidity, but also the textual property of catalysts might have an important effect on the catalytic performance.15–17 In order to verify this speculation, catalysts, S-1, S-2 and S-3, were characterized by TG, XPS, elemental analysis, acid–base titration and N2 adsorption and desorption experiments.
Table 1 The catalytic performance of different catalysts on this reactiona
| Catalyst |
Conversionb (%) |
Selectivityc (%) |
Reaction conditions: temperature: 60 °C; molar ratio of ammonia/acetone: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6; gas hourly space velocity (GHSV): 10.4 h−1. The each data point is an average of three or more runs. The conversion of acetone. The selectivity of triacetonamine. :6; gas hourly space velocity (GHSV): 10.4 h−1. The each data point is an average of three or more runs. The conversion of acetone. The selectivity of triacetonamine. |
| S-1 |
28.7 |
56.2 |
| S-2 |
44.7 |
58.1 |
| S-3 |
35.1 |
57.6 |
| S-4 |
31.1 |
59.9 |
3.2 Catalyst characterization
3.2.1 TG. Thermogravimetric profiles of the three catalysts (Fig. 1) showed similar features, with two distinct weight losses in the ranges of 25–100 °C and 250–750 °C, corresponding to the desorption of physisorbed water and the degradation of the immobilization fraction, respectively. The Fig. 1 clearly displayed that with the increase of MPTMS content, the extent of functionalization increased. However, it was surprised that DTG profiles of these catalysts exhibited two peaks in the range of 300 to 600 °C. The reason may be attributed to the presence of non-oxidized thiol groups, which was further demonstrated by XPS.
 |
| | Fig. 1 TG and DTG profiles of samples S-1, S-2 and S-3. | |
3.2.2 XPS. The sulfur 2p XPS spectrum for catalyst S-2 was shown in Fig. 2. It indicated that there existed two states of sulfur on the surface of catalyst S-2. A binding energy of about 169 eV corresponded to –SO3H and 164 eV corresponded to –SH.18 Although XPS technique was only sensitive to close-to-surface regions of catalysts, the results still gave a beautiful illustration of the two peaks of DTG. Obviously, the total level of functionalization did not reflect the acidity of the catalysts.
 |
| | Fig. 2 XPS spectrum of sample S-2. | |
3.2.3 Physical and textural properties. The carbon-to-sulfur molar ratios were obtained by elemental analysis, which reflected the way of MPTMS immobilization. For the unreacted MPTMS, the C/S molar ratio is six. When one methoxy group reacted with a silanol group, the C/S ratio dropped by one. The obtained results suggested that two or three methoxy groups on MPTMS were condensed with silanol to form oxygen bridges on the silica surface. Meanwhile, the results of elemental analysis also showed the bulk sulfur content of the three catalysts, which reflected the total level of functionalization. The results were in accordance with the TG experiment. The acidity of the three catalysts was determined by acid–base titration and the results (Table 2) indicated that with the increase of MPTMS content, the acidity of catalyst increased from 0.76 to 1.02 mmol g−1. In addition, the N2 adsorption and desorption experiments were performed and the results were shown in Fig. 3. As Fig. 3 displayed, catalyst S-1 exhibited a typical isotherm of mesoporous materials with a hysteresis. However, with the increase of the functionalization level, the hysteresis disappeared gradually and the N2 isotherms changed obviously from type IV to type I. Catalyst S-3 exhibited a typical isotherm of microporous materials. Meanwhile, Table 2 showed that the surface area, the mesopore volume and the average pore diameter decreased sharply as the functionalization level increased. In general, the decreases of the surface area certainly lead to the decreases of catalytic activity. Moreover, the decreases of the mesopore volume and the average pore diameter would respectively bring down the amounts of effective active sites and hinder the diffusion of acetone, which would lead to the decreases of acetone conversion.
Table 2 Physical and textural properties of sample S-1, S-2 and S-3
| Catalyst |
Bulk C/S molar ratio |
Bulk S (m mol g−1) |
Aciditya (mmol g−1) |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Meso pore volumeb (cm3 g−1) |
Average pore diameter (nm) |
| The acidity is determined by acid–base titration. Mesopore volume (Vtotal − Vmicro), Vtotal is the total pore volume tested at p/p0 = 0.99. |
| S-1 |
3.26 |
1.38 |
0.76 |
598 |
0.63 |
0.466 |
4.18 |
| S-2 |
3.67 |
1.84 |
0.98 |
548 |
0.51 |
0.351 |
3.71 |
| S-3 |
3.84 |
2.50 |
1.02 |
464 |
0.30 |
0.123 |
2.57 |
 |
| | Fig. 3 N2 adsorption and desorption isotherms for S-1, S-2 and S-3. | |
3.3 Catalytic activity
With S-2 as the catalyst, the influence of molar ratio, temperature and GHSV on the reaction of acetone and ammonia were investigated. The reaction mixture was analyzed by GC-MS. As expected, mesityl oxide (MO), acetonine (AC), diacetone alcohol (DAA) and diacetone amine (DAM) were detected. In addition, 2,6-dimethylhepta-2,5-dien-4-imine (DMD)19,20 and the high-boiling substances (HBS) were also existed in the reaction mixture. The structures of the aforementioned compounds were described in Fig. 4.
 |
| | Fig. 4 The products of the reaction of acetone and ammonia. | |
3.3.1 The effect of ammonia/acetone molar ratio on the reaction. As the name implies, triacetonamine is obtained from three molecules of acetone and one molecule of ammonia. There is no doubt that the molar ratio of ammonia to acetone plays a crucial role in the synthesis of triacetonamine. Therefore, the influence of ammonia/acetone molar ratio on this reaction was first investigated and the results were shown in Table 3. It could be seen that with the increase of ammonia/acetone molar ratio from 1:12 to 1:6, both of the conversion of acetone and the selectivity of triacetonamine increased. When the molar ratio increased from 1:6 to 1:3, the selectivity of triacetonamine decreased from 58.1% to 44.2%. However, the selectivity of acetonine and the high-boiling substances increased significantly. Therefore, the decrease of triacetonamine selectivity must be mainly attributed to the formation of acetonine and the high-boiling substances.
Table 3 The effect of ammonia/acetone molar ratio on the reactiona
| Entry |
Molar ratio |
Conversionb (%) |
Selectivity (%) |
| MO |
DAA |
DAM |
DMD |
AC |
TAAc |
HBS |
| Reaction conditions: temperature: 60 °C; GHSV: 10.4 h−1. The each data point is an average of three or more runs. The conversion of acetone. Triacetonamine was represented by TAA. |
| 1 |
1:12 |
39.1 |
21.9 |
— |
— |
13.7 |
9.7 |
51.4 |
3.3 |
| 2 |
1:6 |
44.7 |
8.3 |
— |
— |
8.5 |
15.2 |
58.1 |
9.9 |
| 3 |
1:3 |
58.8 |
8.5 |
1.0 |
4.7 |
4.7 |
22.4 |
44.2 |
14.5 |
Diacetone alcohol and diacetone amine were also detected and their selectivities were 1.0% and 4.7% at the ammonia/acetone molar ratio of 1:3, respectively. Diacetone alcohol and diacetone amine were normally concomitant in the synthesis of triacetonamine from acetone and ammonia. In addition, Table 3 indicated that with the increase of ammonia/acetone molar ratio from 1:12 to 1:6, the selectivity of mesityl oxide decreased sharply. The condensation of acetone to mesityl oxide might be the first step in the synthesis of triacetonamine. Once mesityl oxide was generated, it acted as the starting materials to give diacetone alcohol, diacetone amine, acetonine and triacetonamine through a series of reactions.
3.3.2 The effect of temperature on the reaction. The effect of temperature on this reaction was also examined and the results were summarized in Table 4. It was found that when the temperature increased from 50 to 70 °C, the conversion of acetone decreased dramatically and the selectivity of triacetonamine increased from 27.2% to 58.1% and then decreased to 26.3% at 70 °C. However, Table 4 clearly shows that the selectivities of mesityl oxide and 2,6-dimethylhepta-2,5-dien-4-imine were as high as 36.2% and 21.2% at 70 °C, respectively. Obviously, the increase of temperature would result in the decrease of ammonia concentration in liquid phase, which inhibited the equilibrium from shifting right. Therefore, 60 °C may be the optimal reaction temperature.
Table 4 The effect of temperature on the reactiona
| Entry |
Temperature (°C) |
Conversionb (%) |
Selectivity (%) |
| MO |
DAA |
DAM |
DMD |
AC |
TAAc |
HBS |
| Reaction conditions: ammonia/acetone molar ratio: 1:6; GHSV: 10.4 h−1. The each data point is an average of three or more runs. The conversion of acetone. Triacetonamine was represented by TAA. |
| 1 |
50 |
60.2 |
5.4 |
3.8 |
7.3 |
13.2 |
28.3 |
27.2 |
14.8 |
| 2 |
60 |
44.7 |
8.3 |
— |
— |
8.5 |
15.2 |
58.1 |
9.9 |
| 3 |
70 |
9.8 |
36.2 |
— |
— |
21.2 |
11.1 |
26.3 |
5.2 |
3.3.3 The effect of GHSV on the reaction. Gas Hourly Space Velocity (GHSV) is another important parameter for the reaction of acetone and ammonia in a fixed-bed reactor. So the catalytic performance of catalyst S-2 under different GHSV was evaluated and the results were shown in Table 5. There was only trace of triacetonamine detected at the GHSV of 20.7 h−1. However, with the decrease of GHSV from 20.7 to 5.3 h−1, the conversion of acetone increased dramatically to 60.2%. As well known, GHSV has a direct influence on the detention time of reagents and the longer detention time was in favor of the conversion of acetone significantly. Meanwhile, the longer detention time would lead to the formation of diacetone alcohol, diacetone amine and the higher boiling substances, which brought down the selectivity of triacetonamine. So 10.4 h−1 was chosen as the GHSV in this study.
Table 5 The effect of GHSV on the reactiona
| Entry |
GHSV (h−1) |
Conversionb (%) |
Selectivity (%) |
| MO |
DAA |
DAM |
DMD |
AC |
TAAc |
HBS |
| Reaction conditions: temperature: 60 °C; molar ratio of ammonia/acetone: 1:6. The each data point is an average of three or more runs. The conversion of acetone. Triacetonamine was represented by TAA. |
| 1 |
20.7 |
Trace |
— |
— |
— |
— |
— |
Trace |
— |
| 2 |
10.4 |
44.7 |
8.3 |
— |
— |
8.5 |
15.2 |
58.1 |
9.9 |
| 3 |
5.3 |
60.2 |
8.9 |
2.4 |
3.4 |
11.0 |
11.2 |
49.4 |
13.7 |
3.4 Reaction mechanism
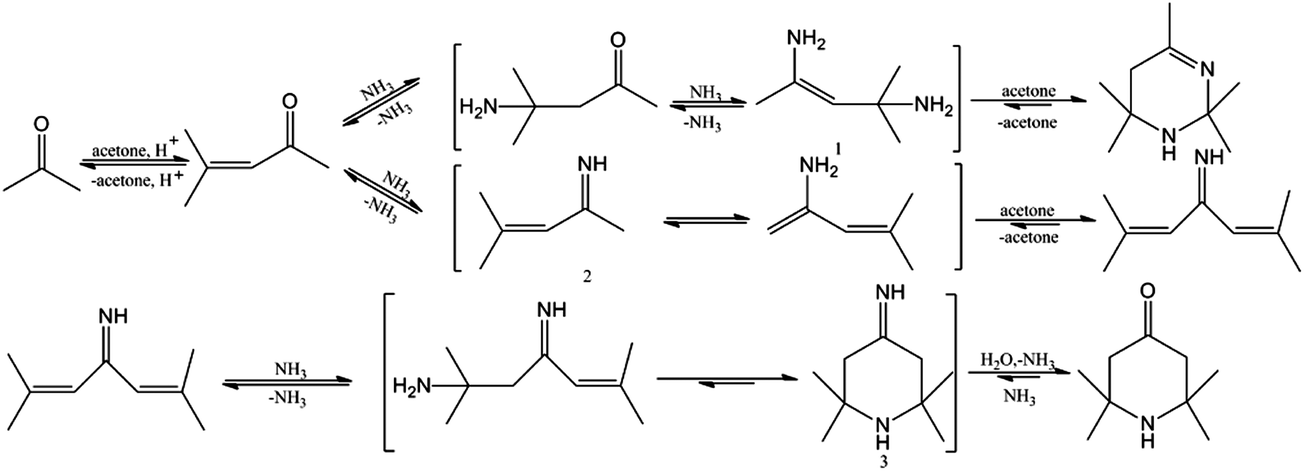
On the basis of the research of Haw et al.19 and Kubelková et al.,20 combined with the experimental results described above, the generation mechanisms of triacetonamine and byproducts were proposed. As shown in Fig. 5, acetone underwent aldol condensation on the acid sites of catalysts to give mesityl oxide, subsequent Michael addition with ammonia to generate diacetone amine. Nucleophilic addition of diacetone amine with ammonia, followed by dehydration afforded diamine intermediate 1. Then, acetonine was formed through nucleophilic addition of diamine intermediate 1 with acetone and following cyclization reaction. In addition, nucleophilic addition and subsequent dehydration of mesityl oxide with ammonia proceed to give imine intermediate 2. Imine intermediate 2 reacted with one molecule of acetone to generate 2,6-dimethylhepta-2,5-dien-4-imine. 2,6-Dimethylhepta-2,5-dien-4-imine underwent Michael addition and intramolecular cyclization with ammonia to yield imine intermediate 3, which was easily converted to triacetonamine by nucleophilic substitution with H2O.
 |
| | Fig. 5 The reaction mechanisms of acetone and ammonia. | |
4. Conclusion
The continuous synthesis of triacetonamine from acetone and ammonia was realized over sulfonic acid-functionalized mesoporous silicas. The catalysts were characterized and the results indicated that with the increase of MPTMS content, the total level of functionalization and the bulk concentration of sulfonic acid increased. However, the surface area, the mesopore volume and the average pore diameter of functionalized catalysts decreased sharply. The decreases of mesopore volume and average pore diameter would respectively reduce the effective active sites and hinder the diffusion of acetone, which brought down the conversion of acetone. Therefore, catalyst S-2 exhibited better catalytic performance. Meanwhile, the influences of molar ratio, temperature and GHSV on the reaction of acetone and ammonia were investigated over catalyst S-2. And an optimized process for synthesizing triacetonamine was obtained. Based on the experimental results above, the reaction mechanisms of acetone and ammonia were speculated.
References
- R. E. Malz Jr, Y.-C. Son and S. L. Suib, Process for the synthesis of 2,2,6,6-tetramethyl-4-oxopiperidine, US 2002/0128482A1, 2002.
- H. K. Hall Jr, Steric effects on the base strengths of cyclic amines, J. Am. Chem. Soc., 1957, 79, 5445–5447 Search PubMed.
- I. Orban, H. Lind and J. Rody. Process for the preparation of 2,2,6,6-tetramethyl-4-oxopiperidine, US 3960875, 1976.
- I. Orban, H. Lind and J. Rody, Process for the preparation of 2,2,6,6-tetramethyl-4-oxopiperidine, US 3953459, 1976.
- I. Orban, J. Rody and M. Rasberger. Process for the preparation of 2,2,6,6-tetramethyl-4-oxopiperidine, US3959295, 1976.
- T. Haruna, A. Nishimura and K. Sugibuchi. Method for preparing 2,26,6-tetramethyl-4-oxopiperidine, US4663459, 1987.
- W. M. Kruse and J. F. Stephen. Process for the perpartion of 2,2,6,6-tetramethyl-4-oxopiperidine, US4734502, 1988.
- Y. Nakahara, Kubota and T. Haruna, Process for preparing triacetoneamine, US4418196, 1983.
- H. Wiezer, G. Nowy and H. Häberlein, Process for the preparation of 2,2,6,6-tetramethylpiperidone-4, US4356308, 1982.
- M. Julius and H. Siegel, Preparation of 2,2,6,6-tetramethylpiperidin-4-one (TAA), US5856494, 1999.
- B. Hirai, N. Kubota and Y. Nakahara. Process for preparing 2,26,6-tetramethyl-4-oxopiperidine, US4252958, 1981.
- Z. Dolejšek, J. Nováková, V. Bosáček and L. Kubelková, Reaction of ammonia with surface species formed from acetone on a HZSM-5 zeolite, Zeolites, 1991, 11, 244–247 CrossRef.
- H. Mochizuki, T. Yokoi and H. Imai, et al., Effect of desilication of HZSM-5 by alkali treatment on catalytic performance in hexane cracking, Appl. Catal., A, 2012, 449, 188–197 CrossRef CAS PubMed.
- Z. Ma, Q. Huang and J. M. Bobbitt, Oxoammonium salts. 5.1 a new synthesis of hindered piperidines leading to unsymmetrical TEMPO-type nitroxides. Synthesis and enantioselective oxidations with chiral nitroxides and chiral oxoammonium salts, J. Org. Chem., 1993, 58, 4837–4843 CrossRef CAS.
- M. L. Testa, V. L. Parola and A. M. Venezia, Transesterification of short chain esters using sulfonic acid-functionalized hybrid silicas: Effect of silica morphology, Catal. Today, 2004, 223, 115–121 CrossRef PubMed.
- H.-Y. Wu, F.-K. Shieh, H.-M. Kao, Y.-W. Chen, J. Rani Deka, S.-H. Liao and K. C. W. Wu, Synthesis, bifunctionalization, and remarkable adsorption performance of benzene-bridged periodic mesoporous organosilicas functionalized with high loadings of aarboxylic acids, Chem. –Eur. J., 2013, 19, 6358–6367 CrossRef CAS PubMed.
- J. Liu, D. Cheng, Y. Liu and Z. Wu, Adsorptive removal of carbon dioxide using polyethyleneimine supported on propanesulfonic-acid-functionalized mesoporous SBA-15, Energy Fuels, 2013, 27, 5416–5422 CAS.
- P. F. Siril, N. R. Shiju, D. R. Brown and K. Wilson, Optimising catalytic properties of supported sulfonic acid catalysts, Appl. Catal., A, 2009, 364, 95–100 CrossRef CAS PubMed.
- T. Xu, J. Zhang and J. F. Haw, Imine chemistry in zeolites: Observation of gem-amino-hydroxy intermediates by in situ 13C and 15N NMR, J. Am. Chem. Soc., 1995, 117, 3171–3178 CrossRef CAS.
- J. Nováková, V. BoSáček, Z. Dolejšek and L. Kubelková, Interaction of acetone with ammonia and alcohols over a HZSM-5 zeolite Part 1. Methanol, J. Mol. Catal., 1993, 78, 43–55 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.