
Green synthesis of reduced graphene oxide/nanopolypyrrole composite: characterization and H2O2 determination in urine†
Abstract
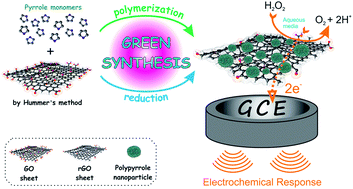
Here we report on a novel, simple and eco-friendly approach for the fabrication of a reduced Graphene Oxide/nanopolypyrrole (rGO/nPPy) composite material and its electrochemical performance for detection of hydrogen peroxide on a glassy carbon electrode. The characterization of the as-prepared rGO/nPPy composite was investigated by Fourier transform infrared spectroscopy, thermogravimetric analysis, ultraviolet-visible spectroscopy, scanning electron microscopy, contact angle measurement, cyclic voltammetry and electrochemical impedance spectroscopy. Cyclic voltammetry, differential pulse voltammetry and chronoamperometry techniques were used to investigate and optimize the performance of the developed electrochemical biosensor. The proposed biosensor showed excellent analytical response towards the quantification of H2O2 at pH 7.40. Under the optimized conditions, the biosensor shows a linear response range from 1.0 × 10−7 to 4.0 × 10−6 M concentrations of H2O2. The limit of detection was determined to be 34 nM. Reproducibility, sensitivity, stability and anti-interference capability of the fabricated biosensor for the detection of H2O2 were examined. The biological relevance of the developed electrochemical biosensor was further studied by the determination of H2O2 in urine samples. The real sample analysis of H2O2 was achieved before and after drinking coffee in urine samples. The successful and sensitive determination of H2O2 urine samples indicates that the proposed electrochemical biosensor can be applied to the quantification analysis of H2O2 in real samples.
 Please wait while we load your content...
Please wait while we load your content...

