 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Prediction of the efficiency of diastereoisomer separation on the basis of the behaviour of enantiomeric mixtures
Emese Pálovicsa,
Zsolt Szeleczkyb,
Balázs Fődib,
Ferenc Faiglab and
Elemér Fogassy*b
aResearch Group for Organic Chemical Technology, University of Technology and Economics, P.O.B. 91 Budapest, Budapest, H-1521, Hungary. E-mail: epalo@mail.bme.hu; Fax: +36 (1)4633648
bDepartment of Organic Chemistry and Technology, University of Technology and Economics, Budapest, Hungary
First published on 10th March 2014
Abstract
The driving force of homo- and heterochiral complex formation in mixtures of chiral compounds is, probably, the effort of the system to separate the most symmetric associates from the less symmetric ones. A possible way to achieve separation of these associates is distribution between two phases. Therefore, during the separation of (a certain part) diastereoisomers similar trends can be observed as in the course of the distribution of enantiomeric mixtures between two phases, although in the first case a third chiral compound (namely the resolving agent) is present. Of course in this case the pursuit of symmetry is not so obvious as in case of enantiomeric mixtures. It should be noted that the outcome may thus be modified by the intervention of kinetic control. One can conclude that the structure of chiral compounds encodes the result of the (optical) resolution.
Introduction
In many cases, living organisms contain only one of the two enantiomers of chiral molecules, but often racemic compounds (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the two enantiomers) are obtained in the chemical syntheses. The biological activity of enantiomers may be different or even opposite, so enantiomeric separations are necessary and inevitable. Many of the methods described in the literature for the separation of enantiomers involve the formation of diastereoisomers followed by liberation of the separated enantiomers. These enantiomeric separation methods are discussed and systematized in several articles.1–10
:1 mixture of the two enantiomers) are obtained in the chemical syntheses. The biological activity of enantiomers may be different or even opposite, so enantiomeric separations are necessary and inevitable. Many of the methods described in the literature for the separation of enantiomers involve the formation of diastereoisomers followed by liberation of the separated enantiomers. These enantiomeric separation methods are discussed and systematized in several articles.1–10
In the course of the resolution processes, racemic compounds are reacted with another chiral reagent (resolving agent). The diastereoisomers so obtained are separated, and their decomposition affords the corresponding enantiomeric mixtures. Usually, pure enantiomers can only be obtained by further purification of these enantiomeric mixtures (Scheme 1).
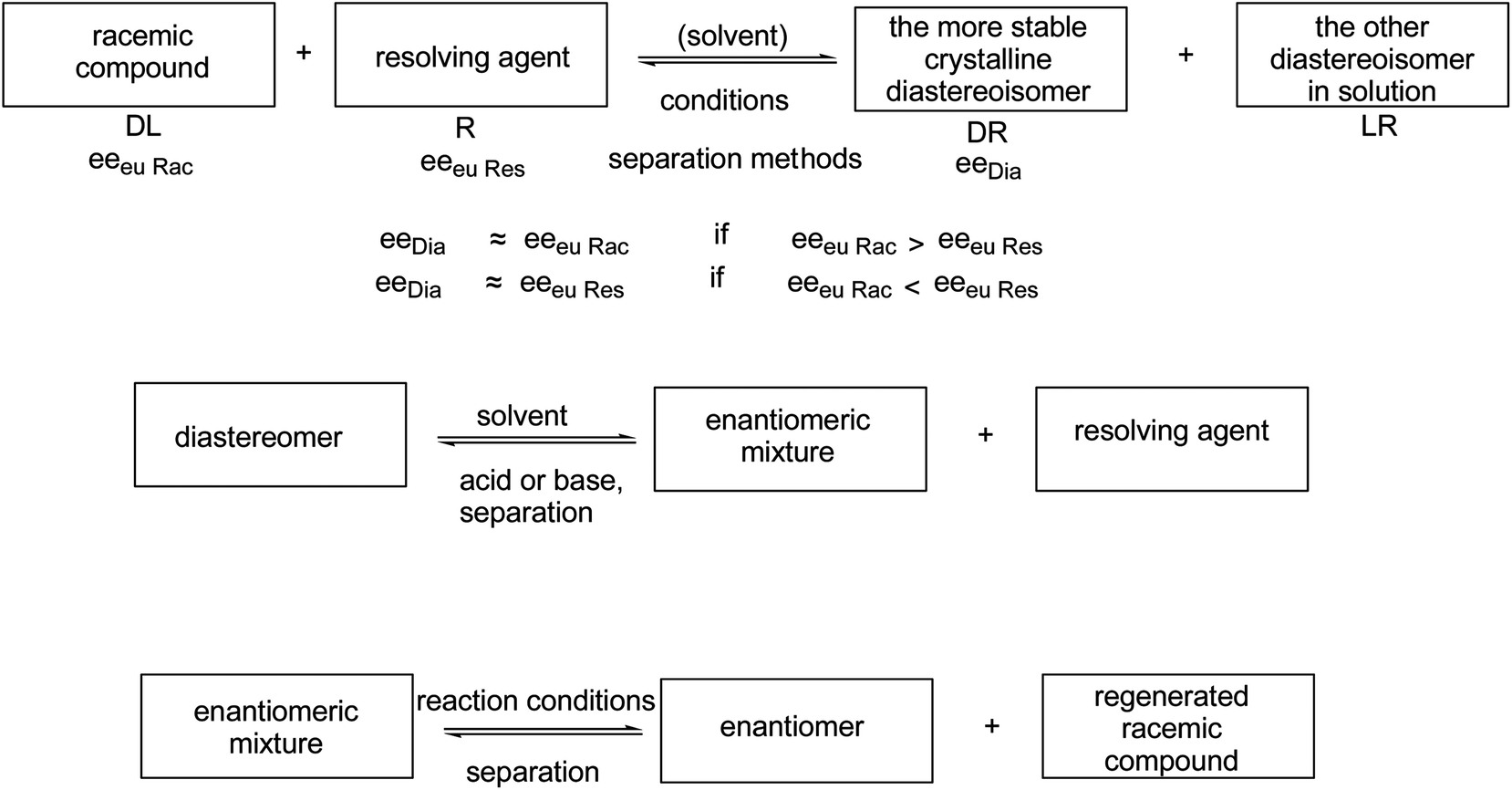 | ||
Scheme 1 General scheme for the preparation of pure enantiomers via diastereoisomeric salt formation (eeeuRac: ee of an enantiomer at the eutectic composition of the racemate; eeeuRes: ee of the resolving agent enantiomer (R or  ) at the eutectic composition; eeDia: ee of the enantiomer of the original racemate at the eutectic composition of the diastereoisomeric salt). ) at the eutectic composition; eeDia: ee of the enantiomer of the original racemate at the eutectic composition of the diastereoisomeric salt). | ||
The composition of the salt crystallized during the fractionated precipitation of the diastereoisomeric salts and enantiomeric mixtures is determined by the SDE11 capacity (self disproportion of enantiomers) of the involved chiral compounds.
The most commonly used methods for the separation of enantiomeric mixtures are based on the exploitation of the distribution of hetero- and homochiral associates between two phases (most often between solid and liquid or vapour phases).12 According to the most recent research enantiomeric enrichment can occur using achiral chromatography13–16 and by separation of racemates using a chiral selector on an achiral17,18 stationary phase or chiral phase chromatography (HPLC),19 respectively.
A correlation between the eutectic composition of binary melting point diagrams of diastereoisomeric mixtures (eeEuDiast) and efficiency of resolution (F) was established by us (Scheme 2).20
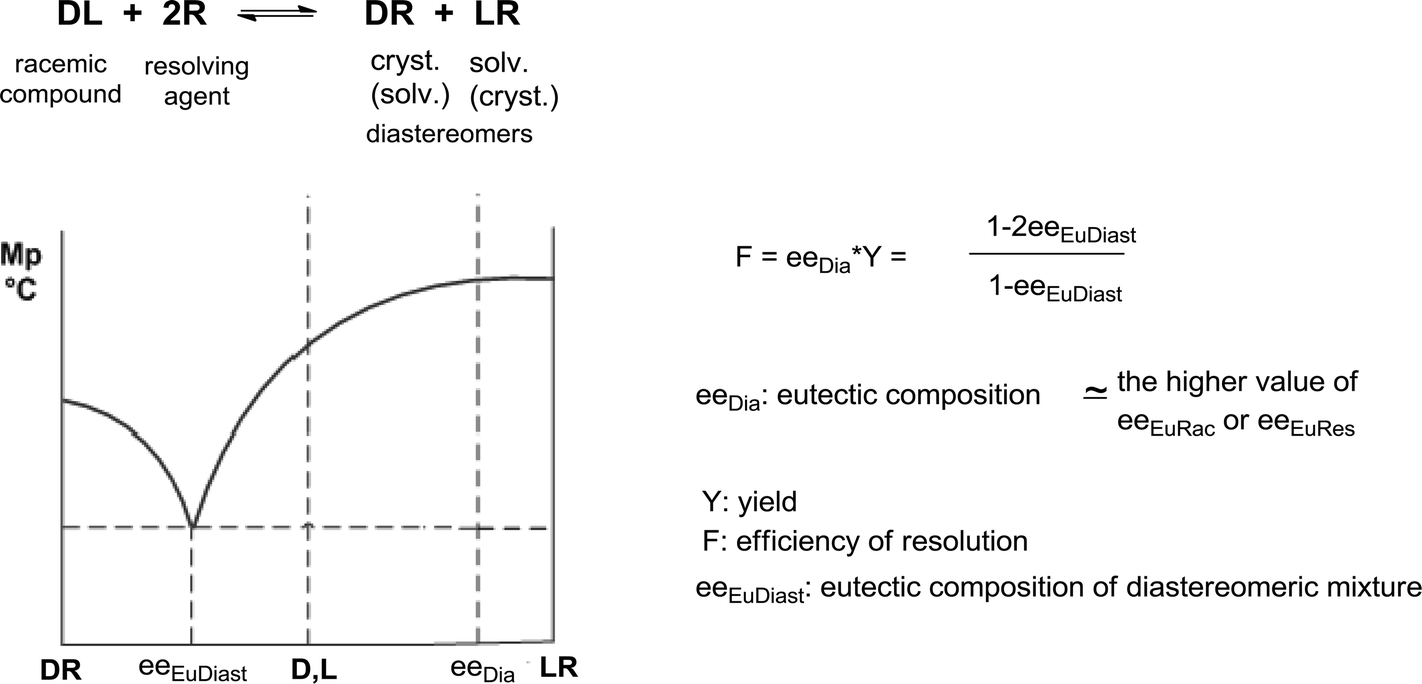 | ||
| Scheme 2 Melting point/composition diagram of diastereoisomers and the relation between eutectic composition of diastereoisomeric mixture and efficiency of resolution. | ||
Furthermore, it was found by analyzing the results of 45 resolutions, that the average enantiomeric purity (eeEuDia = 78%) of enantiomeric mixtures isolated from crystalline diastereoisomers correlates to the average value of the measured eutectic composition of the starting racemic compounds (eeEuRac = 73%). At the same time, when the eutectic composition of the resolving agent is higher than the eutectic composition of the racemic compound (in 29 cases), a better correlation was observed between the average value of enantiomeric purities (eeDia = 80%) of enantiomeric mixtures isolated from the crystalline diastereoisomers and the average value of eutectic compositions of enantiomeric mixtures of the resolving agent (eeEuRes = 78%) (Table 1).21
| Number of resolutions | eeeuRac (average) % | eeeuRes (average) % | eeDia (average) % | F (average) |
|---|---|---|---|---|
| 45 | 73 | — | 78 | 0.57 |
| 29 | — | 78 | 80 | 0.62 |
Based on these observations, we suppose that the composition of a crystalline diastereoisomer is determined either by the eutectic composition of the racemic compound or that of the resolving agent and the higher ee value has a more dominant effect (Scheme 3).
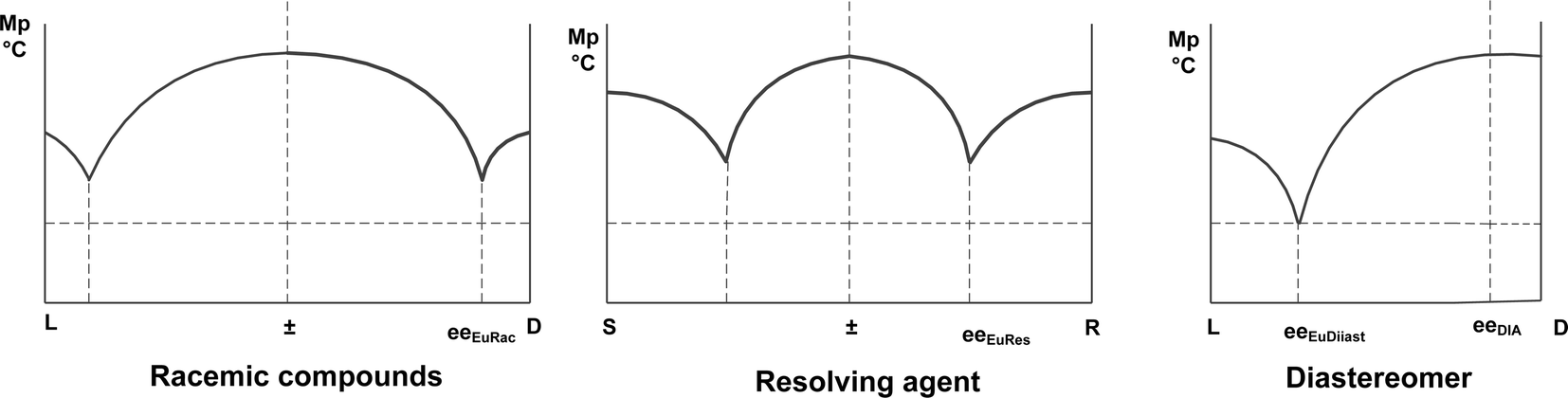 | ||
| Scheme 3 The effect of the eutectic composition of racemic compound and resolving agent on the eutectic composition of diastereoisomers obtained (see also Scheme 2). | ||
Consequently, a correlation can be found between the binary melting point/composition phase diagram of the diastereoisomeric mixtures and the phase diagrams of the enantiomers which are the constituents of the diastereoisomers.
If we wish to separate the enantiomers of a racemic mixture using a structurally similar resolving agent (equivalent or half equivalent amount), the isomers of the racemic compound are transformed into “quasi enantiomeric mixtures”. In the course of separation of these “quasi enantiomeric mixtures” (of diastereoisomers) the same methods based on the distribution between two phases can be used which are suitable methods for the separation of enantiomeric mixtures (also diastereoisomeric related supramolecular enantiomeric associates).7
Separation of diastereoisomers from the melt
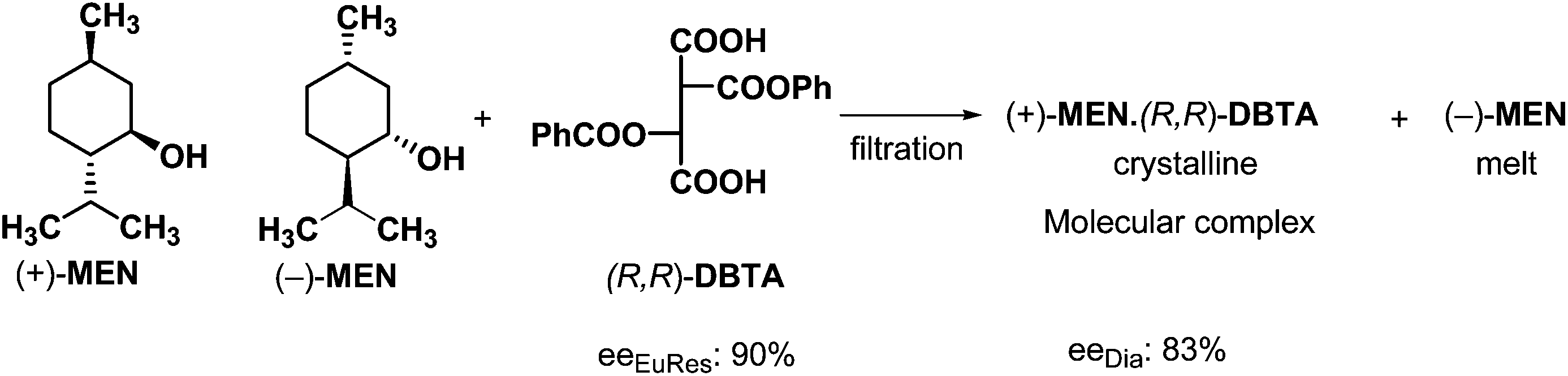
The mixtures of racemic compounds and resolving agents can be considered as mixtures of diastereoisomeric supramolecular structures. These diastereomeric supramolecular structures exist in solutions and in melts, therefore these diastereoisomeric associates can be separated by crystallization from the melt. In this case the mixture of the racemic compound and the resolving agent is melted, then the crystalline phase – obtained by controlled cooling – can be separated by filtration (if it is possible).An example of such a separation is the crystallization of a diastereoisomeric mixture of MEN–DBTA molecular complex from a melt incorporating menthol (MEN) and (R,R)-dibenzoyl-tartaric acid ((R,R)-DBTA) (Scheme 4).22
 | ||
| Scheme 4 Separation from the melt. | ||
It can be seen from the above example that the before mentioned behavior of mixed chiral compounds is valid not only for diastereoisomeric salts but for diastereoisomers in general. During resolution of racemic menthol with (R,R)-DBTA the parts of the diastereoisomeric complexes are kept together by weak second order interactions, only. Formation of the more stable molecular complex that crystallizes more quickly makes the separation possible.
Separation of diastereoisomers by sublimation of enantiomeric mixtures
Enantiomeric separations can be effectuated even if the mixture of diastereoisomers is obtained in a solid–solid reaction. An example is the reaction of solid 2-iodo-trans-cyclohexanol (ICH) and solid (R,R)-DBTA for three months (Scheme 5). It was observed in the course of the fractionated vacuum sublimation23 of the above mentioned mixture of compounds, that one of the enantiomers of ICH sublimated as the first fraction at a relatively low temperature (T1, Scheme 5), and the other ICH enantiomer sublimated at a higher temperature (T2), after thermal decomposition of the molecular complex formed previously in the solid–solid reaction.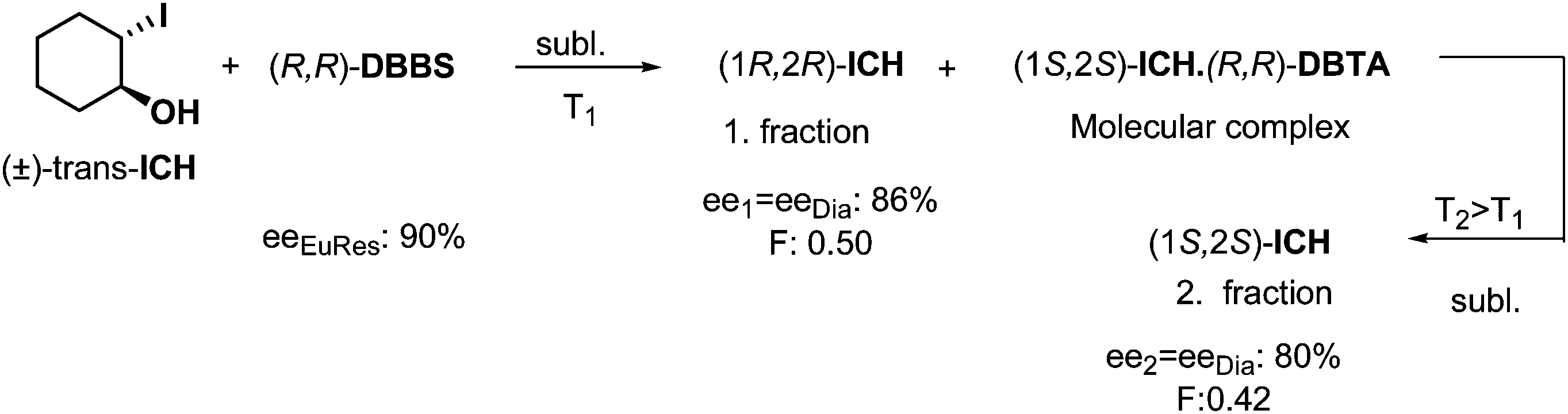 | ||
| Scheme 5 Separation of diastereoisomers by fractionated vacuum sublimation. | ||
Separation of diastereoisomers by distillation of enantiomeric mixtures
In the case of resolutions using half an equivalent of resolving agent it is expected, that the enantiomeric proportion of racemic compound may be separated from the corresponding diastereoisomer formed (distributed between two phases). In the reaction of methylanara (2-methylamino-1-phenylpropane, MA) and (R,R)-DBTA, after the precipitation of the diastereoisomeric salt, the residual free amin, namely (S)-MA could be obtained by distillation under vacuum,24 while the other enantiomer was obtained by the separation of the solid diastereoisomer residue (Scheme 6).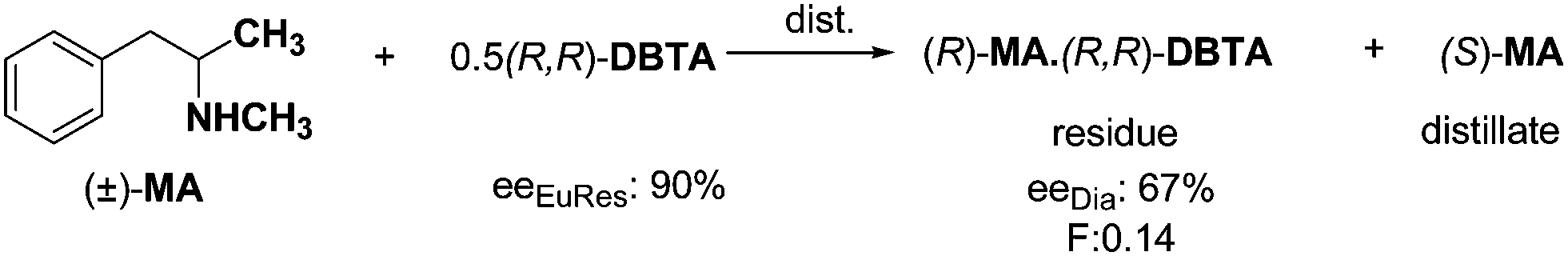 | ||
| Scheme 6 Separation of diastereoisomers by distillation of an enantiomeric mixture. | ||
Separation of diastereoisomers by fractionated distillation of enantiomeric mixtures
This method is also suitable for fractionated separations if the resolving agent forms a salt that can decompose without any damage. An adequate example for this is the resolution of racemic anara (2-amino-1-phenilpropane, AN) by half an equivalent of the structurally related (S)-N-phthaloyl-α-phenylethylamine (PPEA) (Scheme 7). Again, the free, optically active base ((R)-AN) could be distilled off at T1, then the solid diastereoisomeric salt could be decomposed at a higher temperature (T2, by ring closure of the phthaloyl derivative), so the other amine enantiomer ((S)-AN) could be distilled off in the second stage.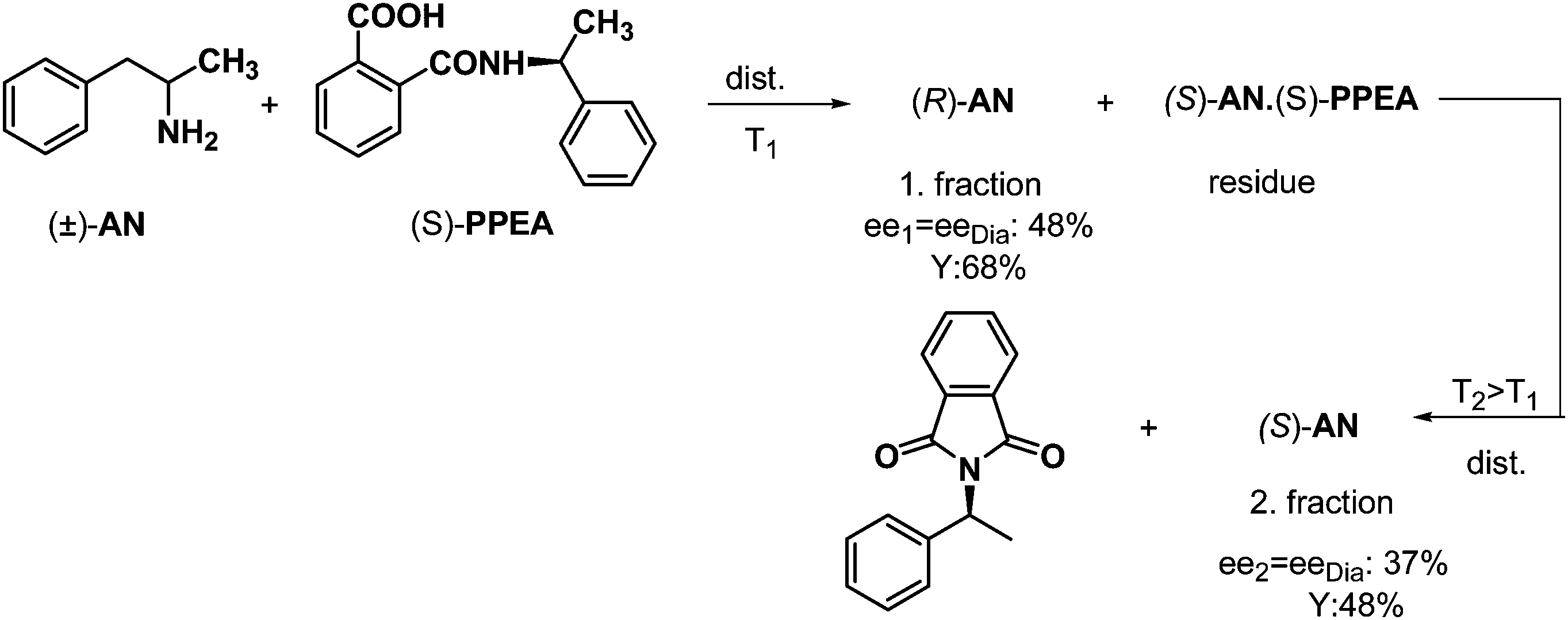 | ||
| Scheme 7 Resolution of racemic AN via fractionated distillation of a mixture of free AN and AN·(S)-PPEA diastereoisomeric salt. | ||
Separation of diastereoisomers by extraction of enantiomeric mixtures with a supercritical fluid (carbon dioxide)
In the course of a half equivalent resolution, the remaining free enantiomer may also be removed by extraction from the reaction mixture after the crystallization of the diastereoisomeric salt. This extraction can be accomplished using a supercritical fluid, most often supercritical carbon dioxide. In case of resolution of trans-cyclohexane-1,2-diol (trans-CHD)25 by (R,R)-tartaric acid ((R,R)-TA) the free enantiomeric portion was separated by extraction with supercritical CO2 from the mixture of the excess of trans-CHD and the crystalline diastereoisomeric complex (Scheme 8). Of course, the other enantiomer can be recovered from the diastereoisomeric complex.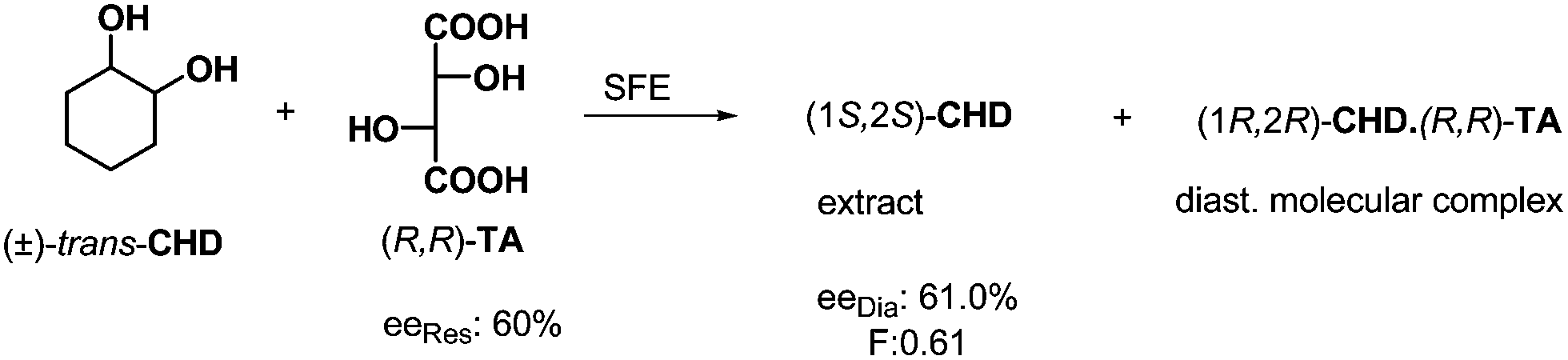 | ||
| Scheme 8 Separation of the trans-CHD enantiomers via molecular complex formation followed with supercritical fluid extraction. | ||
Separation of diastereoisomeric molecular complexes by fractionated crystallization
The above demonstrated methods can be applied for the separation of enantiomers having an asymmetric center on a phosphorous atom. For example, the resolution of several racemic alkyl-, alkoxy-, and aryl-substituted 3-methyl-3-phospholene oxides were accomplished via molecular complex formation with (R,R)-TADDOL (α,α,α′,α′-tetraphenyl-1,3-dioxolan-4,5-dimethanol) as the resolving agent (Scheme 9). If half an equivalent of (R,R)-TADDOL was used, the more stable diastereoisomer crystallized which could be isolated by conventional methods, such as filtration.26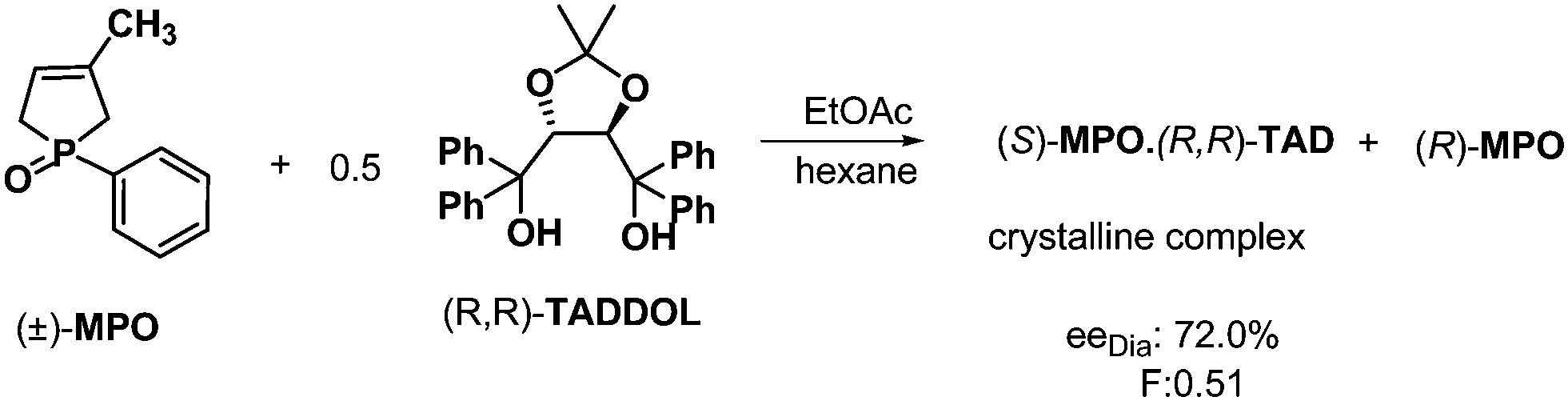 | ||
| Scheme 9 Separation of diastereoisomeric molecular complexes of MPO by fractionated crystallization. | ||
Separation of diastereoisomeric coordination complexes by fractionated crystallization
In other cases, the cyclic P-chiral 3-phospholene oxides were separated into their enantiomeric mixtures by resolution with the Ca2+ or Mg2+ salts of DBTA (DBTC). As is shown in Scheme 10, when half an equivalent of resolving agent was used, the favourable diastereoisomer was precipitated. After filtration and decomplexation of the diastereoisomeric complex, an enantiomeric mixture of the 3-phospholene oxide (MPO) was obtained. The antipode of the first isolated MPO enantiomer was recovered from the mother liquors of the resolutions.27 The effectiveness of the method was demonstrated on a series of cyclic P-chiral 3-phospholene oxides.28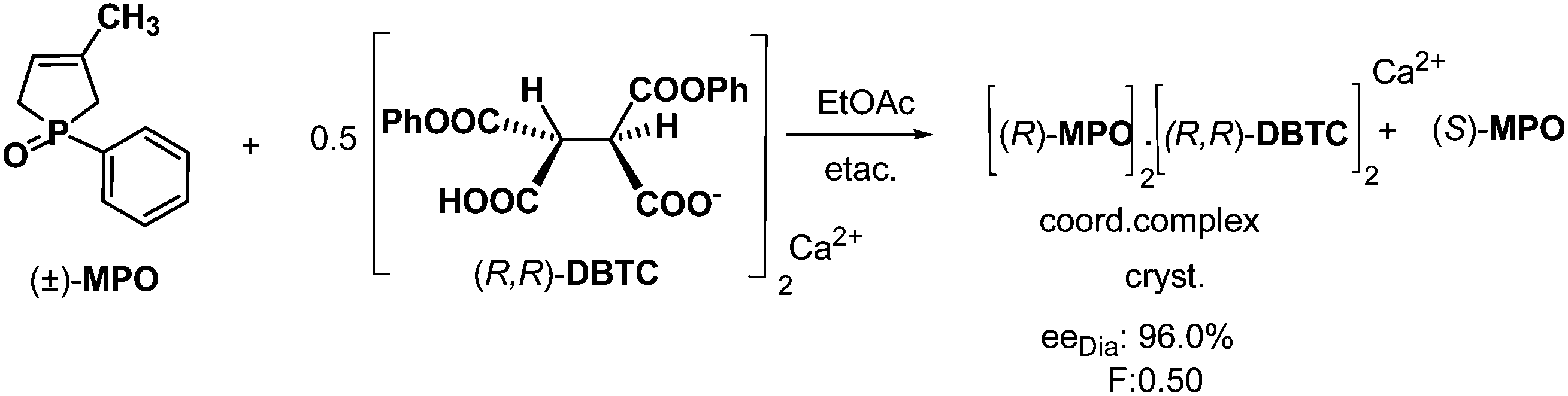 | ||
| Scheme 10 Resolution of MPO via diastereoisomeric coordination complex formation and fractionated crystallization. | ||
Separation of diastereoisomers from enantiomeric mixtures using mixtures of two immiscible solvents
Based on the above examples, formation of solid phase, solid–liquid phases or solid–gas phases systems is always necessary for the separation of diastereoisomers. Now the question is: could one achieve chiral separation between two immiscible liquid phases? The answer yes, of course. Diastereoisomers may be separated by distribution between two liquid phases (Scheme 11).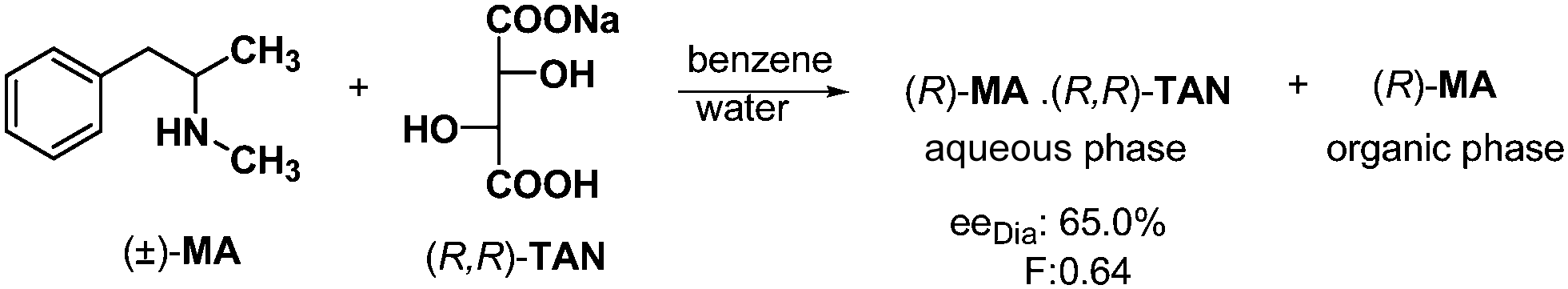 | ||
| Scheme 11 Resolution of MA using two immiscible solvents. | ||
If the racemic methylanara (MA) is reacted with half an equivalent of sodium salt of (R,R)-tartaric acid ((R,R)-TAN) in a mixture of water and benzene, the enantiomer and diastereoisomer are distributed between the two liquid phases.29
The aqueous phase contains the neutral salt of TAN–MA, while the other enantiomer can be found in the organic layer. So the separation of diastereoisomers or enantiomers can be accomplished without crystallization, using two immiscible solvents. The two solvent phases can also provide particularly good separation if the diastereoisomer can crystallize due to its insolubility in the applied two solvents.
Separation of diastereoisomers by crystallization from two immiscible solvents
The resolution of racemic grandaxine (GRA) with half an equivalent of DBTA in a mixture of water and chloroform, or water and dichloromethane is an example of the separation of diastereoisomers by crystallization from two immiscible solvents.30 The crystallization started at the boundary of the solvent phases, then it is accelerated. The formed diastereoisomeric salt was filtrated, and the two phases were separated. The enantiomeric mixture containing one of the enantiomers in excess was obtained from the diastereoisomeric salt, while the other enantiomer was recovered in neutral form from the organic phase. In addition, a small amount of GRA with racemic composition was isolated from the aqueous solution (Scheme 12). From the ee data shown in Scheme 12 one can conclude that in this case, the eutectic composition of the racemic compound governed the efficiency of separation.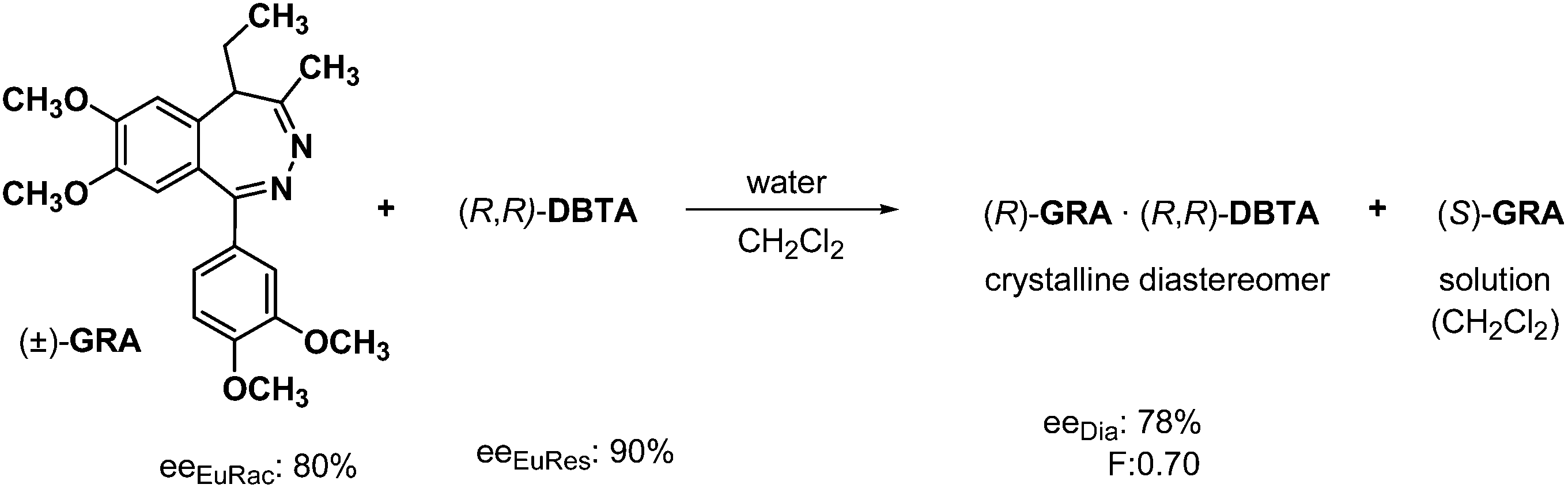 | ||
| Scheme 12 The separation of diastereoisomers by crystallization from immiscible solvents. | ||
Separation of diastereoisomers by crystallization involving the formation of solvates
It is very common during the separation of diastereoisomers, that the enantiomeric purity and yield of separation is increased when solvates were formed during the crystallization of the diastereoisomers. For example, if trans-chrysanthemic acid (CRS) is resolved with N,N-dimethyl-aminodiol (obtained by the transformation of an intermediate of chloramphenicol) in methanol, a methanol solvate of the diastereoisomer could be isolated in very good diastereoisomeric purity, but the yield was 52%, only (Scheme 13). Whereas the resolution was accomplished in diisopropyl ether, or methyl isobutyl ether in the presence of methanol, the methanol solvate of the diastereoisomer was also crystallized, but both the purity and the yield increased significantly.31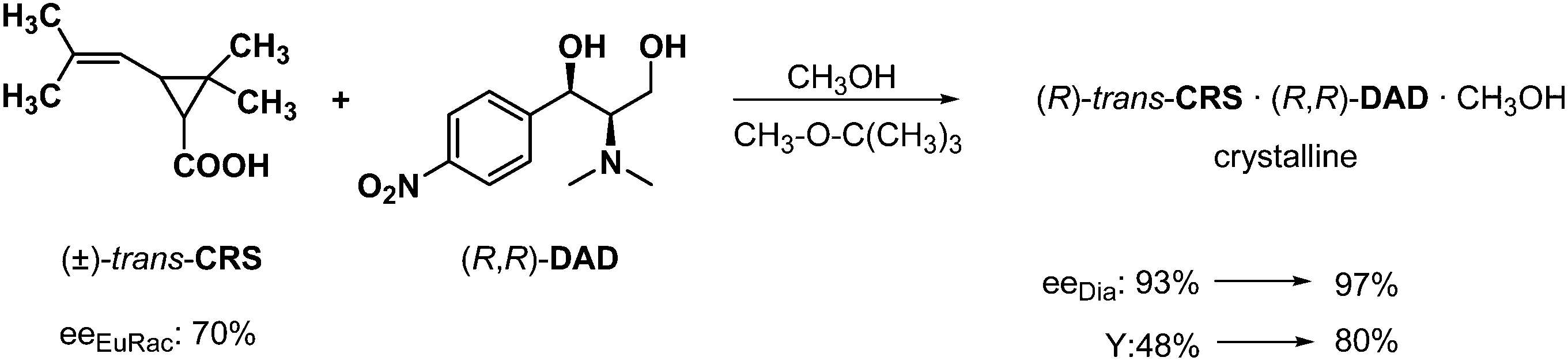 | ||
| Scheme 13 Separation of diastereoisomers by crystallization involving the formation of solvates. | ||
Separation of diastereoisomers by crystallization in the presence of a structurally related achiral reagent
Frequently, the resolution can be accomplished only via solvation. In the previous example the resolution could be carried out using another solvent. When the racemic α-phenylethylamine (PEA) was resolved using half an equivalent of a derivative of a PEA enantiomer, a diastereoisomer was isolated which contained (S)-PEA in 60% excess in acetone (Scheme 14).32 When an achiral compound (such as urea) with a related structure to one part of the resolving agent was added to the solution before crystallization, much purer diastereoisomer was isolated with an (S)-PEA enantiomeric excess of 90%.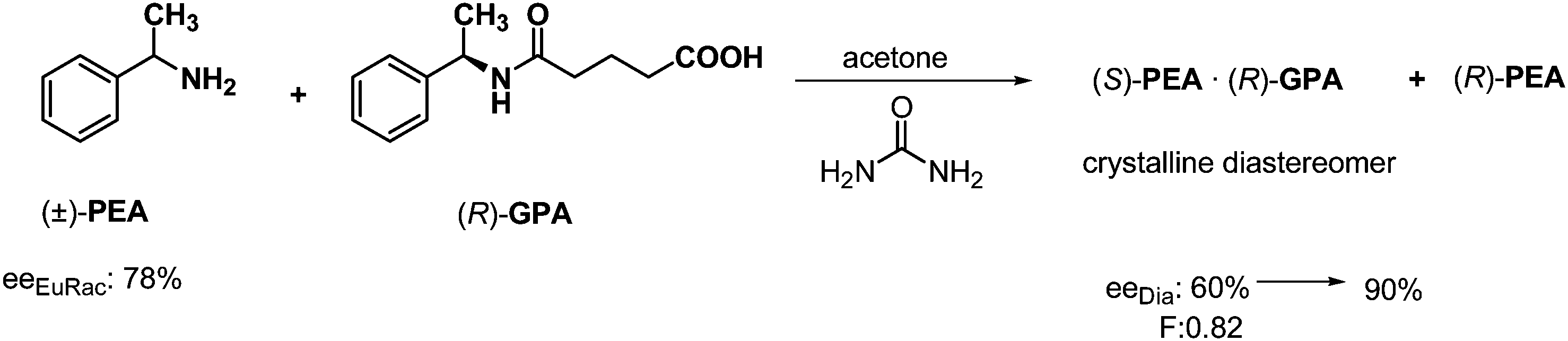 | ||
| Scheme 14 Separation of diastereoisomers by crystallization involving the formation of solvates. | ||
So the achiral solvate forming reagent – which is structurally related to either the resolving agent or the racemic compound – promoted the crystallization of the diastereomer, and increased enantiomeric excess of the diastereomer.
Crystallization of diastereoisomers based on kinetic and thermodynamic control
The decisive role of kinetic and thermodynamic control was observed in the separation of enantiomeric mixtures. This phenomenon can also be found for both the separation of quasi-enantiomeric mixtures and conventional resolutions.33,34 For example, the effect of kinetic control was observed in the resolution of pregabalin (PRE) by mandelic acid (S)-MA (Scheme 15). When the crystalline diastereoisomeric salt was isolated after 15 minutes crystallization, the enantiomeric excess of (S)-PRE isolated from the salt was 98%, while the ee decreased significantly if the crystallization was carried out over 48 hours.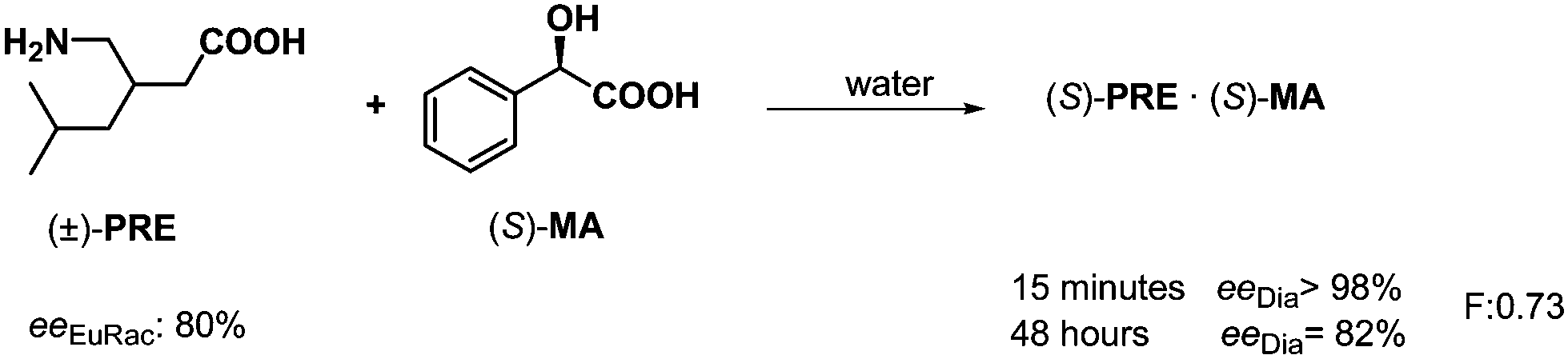 | ||
| Scheme 15 The effect of kinetic control on the separation of diastereoisomers. | ||
This means that the thermodynamic control has a disadvantageous effect on this process. The same phenomenon was observed in the course of the reciprocal process, when racemic MA was resolved by (S)-PRE. Namely, crystallization of the diastereoisomeric salt was controlled kinetically.
In other cases it is necessary to wait until thermodynamic equilibration, because in these cases the process is controlled thermodynamically. An adequate example is the resolution of an intermediate of tamsulozin (TAM) with (R,R)-DBTA (Scheme 16). In this case the diastereoisomeric salt contained the (R)-TAM enantiomer in excess. However, after an hour of crystallization, practically racemic TAM was found in the salt, but excellent enantiomeric excess could be achieved when the diastereoisomer was crystallized for 48 hours.35
 | ||
| Scheme 16 The effect of thermodynamic control on the separation of diastereoisomers. | ||
Conclusion
The above mentioned examples demonstrate that the properties of the involved enantiomeric mixtures determine the enantiomer (diastereoisomer) distribution between two phases and in this way the behaviour of the chiral compounds used determines the efficiency of the process during enantiomer or diastereoisomer separations. There are numerous methods of choice (solid–solid, solid–liquid, solid–gas, liquid–liquid distributions) and, of course, we should choose the most favourable method (if there is more than one possibility).We have recognized that in the resolution processes the diastereoisomers behave similarly to their constituent enantiomeric mixtures if the resolving agent was structurally related to the racemic compound.
We demonstrated that the eutectic composition of the racemate and/or the resolving agent determines the composition of the formed (crystalline) diastereoisomers even if the diastereoisomer forming chiral compounds are not structurally similar. Comparison of the average ee values of the obtained enantiomeric mixtures from series of resolutions with the average eutectic compositions of the involved chiral compounds (racemate and resolving agent) confirmed the above observation, namely the higher eutectic composition governs the enantiomer separation (Table 2).
| No. of experiments | Average value of eeEuRac/eeEuResb | Average value of eeDiab | Average value of F |
|---|---|---|---|
a The used compounds are not structurally related.b eeeuRac: ee of an enantiomer at the eutectic composition of the racemate; eeEuRes: ee of a resolving agent enantiomer (R or 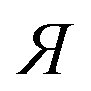 ) at the eutectic composition; eeDia: ee of an enantiomer of the original racemate at the eutectic composition of the diastereoisomeric salt. ) at the eutectic composition; eeDia: ee of an enantiomer of the original racemate at the eutectic composition of the diastereoisomeric salt. |
|||
| 13 (10a) | 80% | 78% | 0.62 |
We also think that the eutectic composition of the diastereoisomer forming enantiomers determines the efficiency of the resolutions in cases of crystalline diastereoisomeric salt, molecular- or coordination complex formations and these governing effects are valid when the separation is based on the distribution between two liquid phases.
Acknowledgements
The authors are grateful for the financial support of the Hungarian Scientific Research Found (OTKA grant number K 104769 for E. Fogassy and K 104528 for F. Faigl) of the New Széchenyi Development Plan (TÁMOP-4.2.1/B-09/1/KMR-2010-0002) and to Gedeon Richter Plc. for the Gedeon Richter PhD Scholarship (Zs. Szeleczky).References
- P. Newman, Optical resolution procedures of chiral compounds 1-3, Resolution Information Center, New York, 1978–1984 Search PubMed.
- S. H. Wilen, Topics in Stereochemistry, ed. E. L. Eliel and N. L. Allinger, Wiley-Interscience, New York. 1972, vol. 6, p. 107 Search PubMed.
- J. Jaques, S. H. Wilen and A. Collet, Enantiomers Racemates and Resolution, Wiley-Interscience, New York, 1981 Search PubMed.
- F. Faigl and D. Kozma, Enantiomer Separation: Fundamentals and Practical Methods, ed. F. Toda, Kluwer Academic Press, Dordrecht, 2004, p. 73 Search PubMed.
- E. Fogassy, M. Nógrádi, E. Pálovics and J. Schindler, Synthesis, 2005, 10, 1555 CrossRef PubMed.
- E. Fogassy, M. Nógrádi, D. Kozma, G. Egri, E. Pálovics and V. Kiss, Org. Biomol. Chem., 2006, 16, 3011 Search PubMed.
- F. Faigl, E. Fogassy, M. Nógrádi, E. Pálovics and J. Schindler, Tetrahedron: Asymmetry, 2008, 4, 519 CrossRef PubMed.
- R. A. Sheldon, Chirotechnology, Marcel Dekker Inc, New York, 1993 Search PubMed.
- S. H. Wilen, A. Collet and J. Jaques, Tetrahedron, 1977, 33, 2725 CrossRef CAS.
- Y. Kobayash, K. Kodama and K. Saigo, Org. Lett., 2004, 17, 2941 CrossRef PubMed.
- V. A. Soloshonok, Angew. Chem., Int. Ed., 2006, 45, 766 CrossRef CAS PubMed.
- F. Faigl, E. Fogassy, M. Nógrádi, E. Pálovics and J. Schindler, Org. Biomol. Chem., 2010, 8, 947–959 CAS.
- J. Martens and R. Bhushan, J. Liq. Chromatogr., 1992, 15, 1–27 CrossRef CAS.
- T. Nakamura, K. Tateishi, S. Tsukagoshi, S. Hashimoto, S. Watanabe, V. A. Soloshonok, J. L. Aceña and O. Kitagawa, Tetrahedron, 2012, 68, 4013–4017 CrossRef CAS PubMed.
- A. E. Sorochinsky, T. Katagiri, T. Ono, A. Wzorek, J. L. Aceña and V. A. Soloshonok, Chirality, 2013, 25, 365–368 CrossRef CAS PubMed.
- V. A. Soloshonok and D. O. Berasov, Chem. Today, 2008, 24, 44 Search PubMed.
- K. Günther, J. Martens and M. Schickedanz, Angew. Chem., Int. Ed. Engl., 1984, 23, 506 CrossRef.
- K. Tateishi, S. Tsukagoshi, T. Nakamura, S. Watanabe, V. A. Soloshonok and O. Kitagawa, Tetrahedron Lett., 2013, 54, 5220–5223 CrossRef CAS PubMed.
- A. M. Krstulović, Chiral Separations by HPLC, Ellis Horwood, 1989, p. 548 Search PubMed.
- D. Kozma, Gy. Pokol and M. Ács, J. Chem. Soc., Perkin Trans. 2, 1992, 435 RSC.
- E. Pálovics, Zs. Szeleczky, F. Faigl and E. Fogassy, New trends and strategies in the chemistry of advanced materials, ed. S. G. Muntean and R. Tudose, 2013, p. 74 Search PubMed.
- K. Simon, Z. Vincze, K. Marthi, G. Lévai, Gy. Pokol, E. Fogassy and D. Kozma, J. Therm. Anal. Calorim., 2004, 75, 787 CrossRef.
- Cs. Kassai, D. Kozma and E. Fogassy, Synth. Commun., 2006, 36, 1015 CrossRef CAS.
- M. Ács, A. Mravik, E. Fogassy and Zs. Böcskei, Chirality, 1994, 6, 314 CrossRef.
- P. Molnár, P. Thörey, Gy. Bánsághi, E. Székely, L. Poppe, A. Tomin, S. Kemény, E. Fogassy and B. Simándi, Tetrahedron: Asymmetry, 2008, 19, 1587 CrossRef PubMed.
- T. Novák, J. Schindler, V. Ujj, M. Czugler, E. Fogassy and Gy. Keglevich, Tetrahedron: Asymmetry, 2006, 17, 2599 CrossRef PubMed.
- V. Ujj, J. Schindler, T. Novák, M. Czugler, E. Fogassy and Gy. Keglevich, Tetrahedron: Asymmetry, 2008, 19, 1973 CrossRef CAS PubMed.
- P. Bagi, E. Fogassy and Gy. Keglevich, Hung. Pat. Appl. 1200228, 2012.
- M. Ács, D. Kozma and E. Fogassy, ACH – Models Chem., 1995, 132, 475 Search PubMed.
- E. Fogassy, G. Tóth, K. Simon, T. Láng, L. Ladányi and L. Párkányi, J. Mol. Struct., 1978, 147, 143 CrossRef.
- M. Ács, D. Kozma, É. Kozsda, Gy. Keserű, Zs. Böcskei, J. Szilágyi, K. Simon, B. Bertók and E. Fogassy, J. Chem. Soc., Perkin Trans. 2, 2000, 149 Search PubMed.
- J. Schindler, M. Egressy, L. Bereczki, Gy. Pokol, E. Fogassy and K. Marthi, Chirality, 2007, 19, 239 CrossRef CAS PubMed.
- J. Neu, E. Fogassy, N. Szalma, P. Kálvin, J. Schindler, G. Jakab, S. Garadnay and E. Pálovics, Hung. Pat. Appl. 1000186, 2010.
- E. Fogassy, E. Pálovics and Zs. Szeleczky, Hung. Pat. Appl. 1200227, 2012.
- T. Gizur, J. Törley, E. Fogassy, G. Egri, J. Bálint and Á. Demeter, Hung. Pat. 202963, 2002.
| This journal is © The Royal Society of Chemistry 2014 |