DOI:
10.1039/C4RA00484A
(Paper)
RSC Adv., 2014,
4, 12710-12715
Mechanical properties of PGA at different water fractions – a molecular dynamics study
Received
17th January 2014
, Accepted 13th February 2014
First published on 14th February 2014
Abstract
The mechanical properties of polyglycolic acid (PGA) of different water weight fractions (1.7%, 2.9%, and 5%) were investigated by molecular dynamics (MD) simulation through a tensile test. The variation in the degree of crystallinity with water content was also investigated using XRD profiles. The Young's modulus, mechanical strength, and fracture mechanism of all PGA–water systems were drawn from the corresponding stress–strain profiles. Furthermore, water diffusion behavior within the PGA matrix both before and after the tension test was also studied. The water diffusion coefficients were derived from the mean square displacements of all water molecules within the PGA matrix.
1 Introduction
In recent years, natural or synthetic polymers with biologically decomposable characteristics have been extensively used in physical and cosmetic surgery as surgical sutures, vascular stents,1 and cartilage supports.2–11 Such materials will gradually break down and decompose after the parts they support within the human body gradually recover to their original functions. It has been proven they are almost harmless and cause nearly no side effects in the human body.12 Another reason to use these materials is for ecological protection by reducing the use of non-environmentally friendly substances, which degrade over hundreds of years.13 The decomposition of biodegradable polymers can be attributed to heat,14 oxidation, photolysis and radiation or hydrolysis. During the decomposition process, the polymer main chain or branched chain is disconnected, and its molecular weight will decrease. For some cases, the original polymer even converts into other molecules.14
Starting several decades ago, these biodegradable polymers began to be used inside the human body for medical treatments.15 PGA is exposed to physiological conditions; it will be randomly degraded by hydrolysis into shorter PGA fragments. Glycolic acid, which is nontoxic to the human body, is the final degradation product. It can easily go through the tricarboxylic acid cycle and be removed as water and carbon dioxide from the human body by breathing. Part of the glycolic acid can be also excreted via kidney and urine.16 PGA exhibits a degree of crystallinity around 46–52% (ref. 17) and has a glass transition temperature (Tg) between 35 and 40 °C,18 resulting in good mechanical properties with flexibility when used as a biomaterial inside the human body. Because of its high degree of crystallisation, it is not soluble in most organic solvents; the exceptions are highly fluorinated organics such as hexafluoroisopropanol. In Montes' study,19 they investigated the crystallinity of PGA materials from different annealing processes and found that the Tg value and crystallinity of PGA can be adjusted by thermal treatment, and the time period of PGA degradation can also be controlled, which makes the application of PGA broader.
The strength of shearing, tension and compression of PGA are usually adopted to observe its mechanical properties because Young's modulus, bulk modulus, and shear modulus20 are important factors influencing degradation time. Since direct investigation of molecular behaviour of PGA under mechanical deformation is difficult due to the very small scales involved, numerical methods are generally preferred. These methods can assist in clarifying the physical insights gained from experimental results. Furthermore, they can be used to predict possible results in order to obtain a preliminary analysis of the influence of process parameters prior to conducting an expensive experiment. Molecular dynamics (MD) simulation is a powerful tool for the investigation of molecular behaviour at an atomic level. Adopting this technique, Sekine conducted a PGA fiber tensile simulation to monitor the variation of PGA chain conformation.21 They found the PGA torsion angles distribute within a small range after the PGA molecules were stretched to a certain degree of elongation. In Ding's study,22 both molecular dynamics and Monte Carlo method were conducted to simulate the PGA system under tension by using a united-atom potential. They found the Young's modulus of both amorphous PGA and those with partial crystallinity become smaller at higher temperatures (Table 1).
Table 1 Density parameters of different water contents (PGA density experimental values: 1.53; molecular weight (M.W.) of one chain: 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 602 (kg mol−1))
602 (kg mol−1))
| Water contents (wt%) |
H2O (M.W.) |
PGA chains |
Atoms |
Density (kg m−3) |
| 0% |
0(0) |
7 |
8414 |
1.4842 |
| 1.7% |
80(1440) |
7 |
8661 |
1.492 |
| 2.9% |
135(2430) |
7 |
8826 |
1.461 |
| 5% |
240(4320) |
7 |
9141 |
1.465 |
Hurrell et al. found that decomposition behaviour of PGA was active after absorbing water molecules, and mass decreases quickly with an increase in the absorption capacity.23 You et al.24 demonstrate by DSC and WAXD analysis that the crystallinity increases significantly in the initial period of PGA decomposition, and gradually decrease in the later period. In particular, Lyu et al.25 investigated the decomposition process of PLA, which is separated into four stages. They found that the degradation of PLA must pass the water saturation process in the first stage. These studies demonstrate the presence of water absorption and the increase of PGA crystallinity in the decomposition reaction. Since PGA is generally applied in the production of surgical sutures, cartilage, bone screws, and other biomaterials, the different stresses and effect of water are crucial considerations. Therefore, the stress and diffusion behaviours are here investigated at different fractions of water molecules in the PGA system. Furthermore, the amount of crystallinity at different stress levels in the decomposition of PGA is elucidated by XRD analysis.
In previous studies, only experimental results were used to determine mechanical properties. However, the detailed changes of microstructures are difficult to observe. In this work, MD simulation was proposed to simulate the PGA strength variation during the degradation process at different temperatures. The Young's modulus, mechanical strength, and fracture mechanism of all PGA–water systems were obtained by the corresponding stress–strain profiles. Furthermore, the water diffusion behaviours within PGA matrix were also studied.
2 Simulation model
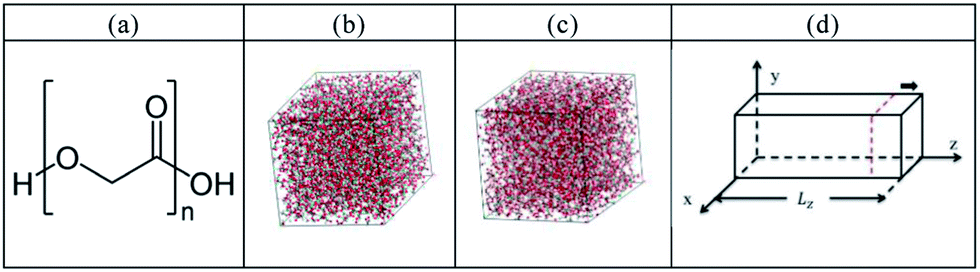
The molecular dynamics simulation was carried out by using the Forcite package,26 and the PCFF force field was used in our simulation with time step of 1 fs for the trajectory integration. In the experiments, water swollen systems for PGA are those at 0%, as well as those corresponding to water content when degradation was detected experimentally (PGA–1.7% water) and when weight loss was detected (PGA–2.9% water). Accordingly, we set up the PGA 200 monomer system with 1.7% and 2.9% water, with one chain including 1202 atoms based on the chemical formula of the monomer shown in Fig. 1(a). In order to better simulate the case of PGA in water, our procedure to obtain the equilibrium configurations of the PGA systems is described as follows: (1) the water and PGA chains were randomly distributed in a large simulation box. (2) After optimization by the conjugate gradient algorithm,27 the system was quenched from 1000 to 300 K with a cooling rate of 10 K for 5000 fs at 1 atm by an isobaric–isothermal ensemble (NPT). The rescaling method28 and the Anderson method29 were used as the thermostat and barostat. (3) Finally, the MD simulation at 298 K and 1 atm was performed for an additional 50 ps by the NPT ensemble to reach the equilibrium condition. The equilibrium configurations of the pure PGA are shown in Fig. 1(b). For the PGA–water system, the configurations are also constructed by the same procedure, and the polymer and water composite material is shown in Fig. 1(c).
 |
| | Fig. 1 (a) PGA chemical formula. Equilibrium configurations of (b) pure PGA and (c) polymer and water composite material. (d) Uniaxial tension model. | |
For the uniaxial tension, the model was then relaxed at 300 K and 1 atm for another 50 ps and this model can be seen in Fig. 1(d). During the tensile process, the cell length in the z-dimension was elongated by 0.25 Å, and then the system was relaxed for 2 ps before imposing the next tensile increment. During the tension simulation, the stress σmn on the m plane and in the n-direction is calculated by30
| |
 | (1) |
where
mi is the mass of atom
i,
Vs is the system volume,
rij is the distance between atoms
i and
j; and
rmij and
rnij are two components of the vector from atoms
i to
j.
Furthermore, the normal strain in the axial direction ε of the PGA is calculated as
| |
 | (2) |
where
lz(t) is the average length in the axial direction and
lz(0) is the initial length of system in the axial direction. Thus, the stress–strain relationship of the PGA can be obtained using
eqn (1) and
(2).
In the crystallinity analysis, X-ray diffraction (XRD) is commonly used as direct evidence of the periodic crystal structure by the relative diffraction intensity at different X-ray incident angles. All XRD profiles shown in the current study were conducted by the REFLEX module in Materials Studio package.31 In REFLEX, Bragg's law is used to obtain the constructive interference intensity for X-rays scattered by materials, and the formula is listed as follows:
| |
 | (3) |
where
θ is a certain angle of incidence when the cleavage faces of crystals appear to reflect X-ray beams.
d is the distance between atomic layers in a crystal, and
λ is the wavelength of the incident X-ray beam. When
n is an integer, the diffraction is constructive with a higher intensity. While
n is an half integer, the diffraction is destructive and the intensity approaches zero.
We calculate the X-ray diffraction patterns after the equilibrium configurations of the procedure. In the calculations, the diffract meter range 2θ was set from 1° to 45° with the step size of 0.05 degree. The X-ray source anode is copper, and the radiation wavelength is set to 1.540562 Å. We calculate the degree of crystallinity after comparing the experimental XRD patterns with the patterns of the structure generated from the MD simulations.
For the degree of crystallinity calculations, the mixture diffraction pattern is composed of the crystalline phase, the amorphous phase and a background contribution. The intensity of input mixture diffraction pattern can be defined as:
| | |
Im(2θ) = Icr(2θ) + Iam(2θ) + Ib(2θ)
| (4) |
where
Icr,
Iam and
Ib, are the sums of the scattering intensities from the crystalline, amorphous phase, and background contribution, respectively.
To obtain the relative weight of the crystalline phase in the decomposition of the mixture diffraction pattern, we can calculate the degree of crystallinity (xcr) from quantitative phase analysis (QPA) theory. This can be defined as the scattering intensity of a powder sample of a single pure phase:
| | |
Icr(2θ) = pcrInormcr(2θ)
| (5) |
| | |
Iam(2θ) = pamInormam(2θ)
| (6) |
| |
 | (7) |
where
pcr is intensity factors, and
Xcr describes crystalline regions (
pcr) and amorphous regions (
pam) of materials in the weight of the total value.
3 Results and discussion
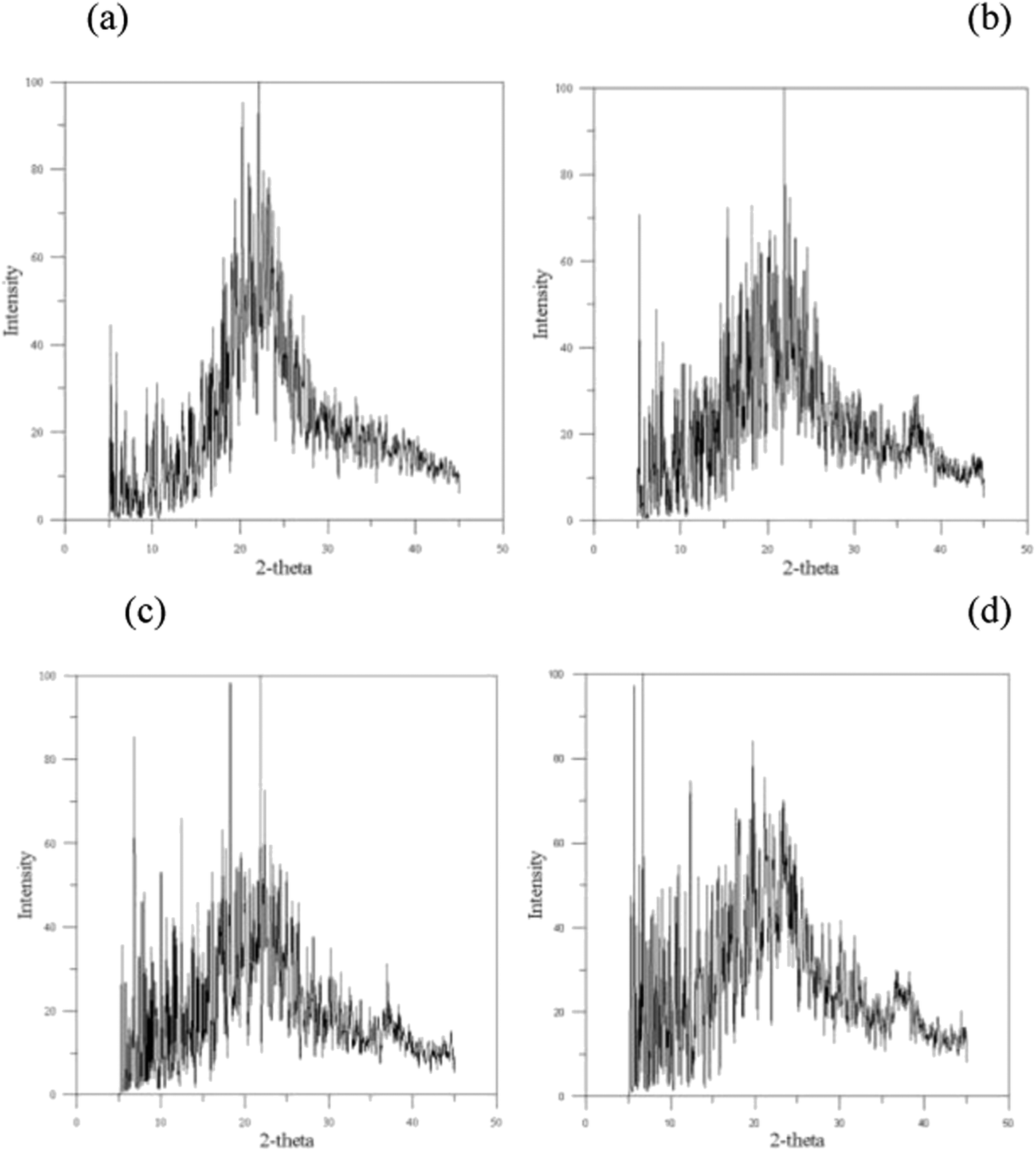
The X-ray diffraction (XRD) profiles for PGA and PGA with water contents of 1.7%, 2.9%, and 5% are displayed in Fig. 2(a)–(d), where no peak for the specific crystalline planes is found in these XRD profiles. However, previous studies of PGA crystallization indicate that PGA displays a partially-crystalline property with the degree of crystallinity increasing with water content until a critical water content.24 In the current cases, the crystallizations of PGA with different water contents do not lead to a 100% pure single crystalline phase, and they result in mixtures of different crystalline and amorphous phases. Accordingly, the XRD profiles shown in Fig. 2(a)–(d) can be used to identify the degree of PGA crystallinity. The XRD profiles require decomposition into three scattering contributions: crystalline, amorphous, and background. The area difference between the crystalline and amorphous contributions determines the degree of crystallinity, and the profile of degree of PGA crystallinity is shown in Fig. 3. The degree of crystallinity of PGA is about 46.7% at PGA-0%, which is located within the experimental observation range of 46% to 52%.17 This result reveals the structure from the annealing process with the PCFF force field can accurately represent PGA material. When the water content increases from 0 to 1.7%, degree of crystallinity rises from 46.7% to 48.2%, only a small increase. At a water content of 2.9%, the degree of crystallinity reaches its maximal value of about 54.5%, and then decreases when the water content exceeds 2.9%. In Hurrell's study,23 they indicated the degree of crystallinity of PGA will decrease when the water content is over 2.9% because these water molecules will enhance the occurrence of hydrolysis and decrease the degree of crystallinity. The experimental critical water content for decreasing the degree of crystallinity of PGA is the same as our MD simulation result.
 |
| | Fig. 2 The X-ray diffraction (XRD) profiles: (a) 0% (b) 1.7% (c) 2.9% (d) 5%. | |
 |
| | Fig. 3 The profile of degree of PGA crystallinity. | |
Fig. 4 shows the mean-square displacement (MSD) plots of pure water and water molecules within PGA at different water contents. The MSD is defined as:
| |
 | (8) |
where
ri(
t) represents the position of the mass centre of water molecule
i at the delay time
t, and
ri(
t0) indicates the referenced position of the corresponding mass centre of water molecule
i at referenced time
t0;
N represents the total number of water molecules within the PGA matrix. The brackets are interpreted as the average over time origins and numbers of atoms. The diffusion of the water molecule was further examined by calculating a self-diffusion coefficient
D from the MSD. The self-diffusion coefficient is obtained from the MSD
via the Einstein equation,
32 which is rewritten as
| |
 | (9) |
 |
| | Fig. 4 The mean-square displacement (MSD) plots of water molecules within PGA at water contents of 1.7%, 2.9%, and 5%. | |
In order to use the optimal simulation parameters for Forcite package on the water diffusion behaviour, the diffusion coefficient of bulk water at 300 K was calculated first in the NVT ensemble by eqn (10) with time step length of 0.4 fs. The value of 2.35 × 10−9 m2 s−1 from our MD simulation is very close to the experimental value of 2.3 × 10−9 m2 s−1,33 indicating the reliability of the PCFF force field to predict the diffusion behaviour of water molecules. The water diffusion coefficients within PGA by eqn (8) are listed in Table 2 for water contents of 1.7%, 2.9%, and 5%.
Table 2 The water diffusion coefficients within PGA
| Water contents |
Diffusion coefficients (m2 s−1) |
| 1.7% |
2.38 × 10−10 |
| 2.9% |
3.46 × 10−10 |
| 5% |
3.07 × 10−10 |
| Pure water32 |
2.3 × 10−9 |
From Fig. 4, it is clear that the MSD value of water content of 2.9% is the largest and the MSD value of 5% water content is smaller than that of 2.9% water content, but larger than that of 1.7% water content. From Fig. 3 and 4, one can see the influence that degree of PGA crystallinity has influence on water diffusion behaviour, such that PGA with a higher degree of crystallinity has a larger water diffusion coefficient. In the comparison between pure water and water-content PGA, the diffusion coefficient of the pure water is significantly larger than that of water within the PGA, indicating the flowed difficulty of water in the polymer structure.
The stress–strain profiles for PGA with 2.9% and 5% water contents under tension are shown in Fig. 5, and the corresponding morphologies at different strains, labelled by (a)–(d) in Fig. 5, are displayed in Fig. 6(a)–(d) for PGA with 5% water content. The stress values for both plots display an abrupt linear increase with strain from 0 to 0.02, indicating an elastic characteristic over this strain range. At strain between 0.02 and 0.2, the stresses fluctuate around a constant value and then display a decreasing trend when the strain continuously increases. In Fig. 6(c), for the morphology at strain of 0.3, there are many voids that have appeared within the PGA matrix and lead to a decrease in stress. In Fig. 6(d), the areas of voids have further expanded, causing the fracture of PGA material. It should be noted the yielding stress is larger than 20 GPa, which is much larger than experimental values. The main reason is the PBC models used in our current study lack free surfaces because of the limitation of computational power. Although the smaller model yields much higher yielding stress for PGA, the tension deformation characteristics obtained are still reasonable when compared to experimental observation.
 |
| | Fig. 5 The stress–strain profiles for PGA with 2.9% and 5% water content under tension. | |
 |
| | Fig. 6 The deformation of PGA–5% water during tension at strains of: (a) 0 (b) 0.1 (c) 0.3 (d) 0.4. | |
4 Conclusions
This study has used MD simulation to investigate the degree of crystallinity of PGA and PGA with 0%, 1.7%, 2.9%, and 5% water content, as well as the diffusion behaviours of water molecules. An increase in water content up to 2.9% will increase the degree of crystallinity of PGA, with the water molecules within the PGA matrix of higher degrees of crystallinity having higher water diffusivity. The stress–strain profiles and the corresponding morphologies at different strains confirm that the expansion of the void areas causes the fracture of PGA at larger strain. The first stage for polyester material is the water saturation process, where the water molecules diffuse into the polyester material, and the results of this study suggest the water diffusion behaviour in this first stage of polyester hydrolysis.
Acknowledgements
Shin-Pon Ju would like to acknowledge the (1) National Science Council, Republic of China, under Grant Number NSC 101-2628-E-110-003-MY3 for the financial support, (2) National Center for High-performance Computing, Taiwan, for the use of computer time, (3) National Center for Theoretical Sciences, Taiwan.
Notes and references
- A. M. Reed and D. K. Gilding, Polymer, 1981, 22, 494–498 CrossRef CAS.
- L. E. Freed, D. A. Grande, Z. Lingbin, J. Emmanual, J. C. Marquis and R. Langer, J. Biomed. Mater. Res., 1994, 28, 891–899 CrossRef CAS PubMed.
- J. Shalhoub, A. Thapar and A. H. Davies, Vascular and Endovascular Surgery, 2011, 45, 422–425 CrossRef PubMed.
- A. A. Haroun, J. Appl. Polym. Sci., 2010, 115, 3230–3237 CrossRef CAS.
- S. Nsereko and M. Amiji, Biomaterials, 2002, 23, 2723–2731 CrossRef CAS.
- T. Bourtoom and M. S. Chinnan, LWT–Food Sci. Technol., 2008, 41, 1633–1641 CrossRef CAS PubMed.
- V. R. Sinha and A. Trehan, J. Controlled Release, 2003, 90, 261–280 CrossRef CAS.
- M. Unverdorben, A. Spielberger, M. Schywalsky, D. Labahn, S. Hartwig, M. Schneider, D. Lootz, D. Behrend, K. Schmitz, R. Degenhardt, M. Schaldach and C. Vallbracht, CardioVascular and Interventional Radiology, 2002, 25, 127–132 CrossRef CAS PubMed.
- D. E. Cutright and E. E. Hunsuck, Oral Surg., Oral Med., Oral Pathol., 1972, 33, 28–34 CrossRef CAS.
- M. L. Cooper, J. F. Hansbrough, R. L. Spielvogel, R. Cohen, R. L. Bartel and G. Naughton, Biomaterials, 1991, 12, 243–248 CrossRef CAS.
- A. Keller, Compos. Sci. Technol., 2003, 63, 1307–1316 CrossRef CAS.
- Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef CAS.
- G. Scott, Polym. Degrad. Stab., 1990, 29, 135–154 CrossRef CAS.
- R. Chandra and R. Rustgi, Prog. Polym. Sci., 1998, 23, 1273–1335 CrossRef CAS.
- H. M. de Oca, D. F. Farrar and I. M. Ward, Acta Biomater., 2011, 7, 1535–1541 CrossRef PubMed.
- D. W. Fry and K. E. Richardson, Biochim. Biophys. Acta, Enzymol., 1979, 567, 482–491 CrossRef CAS.
- H. M. de Oca, I. M. Ward, R. A. Chivers and D. F. Farrar, J. Appl. Polym. Sci., 2009, 111, 1013–1018 CrossRef.
- P. U. Rokkanen, Ann. Med., 1991, 23, 109–115 CrossRef CAS.
- H. Montes de Oca and I. M. Ward, Polymer, 2006, 47, 7070–7077 CrossRef CAS PubMed.
- H.-y. Cheung, M.-p. Ho, K.-t. Lau, F. Cardona and D. Hui, Composites, Part B, 2009, 40, 655–663 CrossRef PubMed.
- S. Sekine, K. Yamauchi, A. Aoki and T. Asakura, Polymer, 2009, 50, 6083–6090 CrossRef CAS PubMed.
- L. Ding, R. L. Davidchack and J. Pan, J. Mech. Behav. Biomed. Mater., 2012, 5, 224–230 CrossRef CAS PubMed.
- S. Hurrell and R. E. Cameron, Biomaterials, 2002, 23, 2401–2409 CrossRef CAS.
- Y. You, B.-M. Min, S. J. Lee, T. S. Lee and W. H. Park, J. Appl. Polym. Sci., 2005, 95, 193–200 CrossRef CAS.
- S. Lyu, J. Schley, B. Loy, D. Lind, C. Hobot, R. Sparer and D. Untereker, Biomacromolecules, 2007, 8, 2301–2310 CrossRef CAS PubMed.
- A. S. Accelrys Materials Studio 5.0, Inc., San Diego, 2010.
- R. Fletcher and C. M. Reeves, Comput. J., 1964, 7, 149–154 CrossRef.
- H. C. Andersen, J. Comput. Phys., 1983, 52, 23–24 CrossRef.
- B. J. Olle Telemana and S. Engströmb, Mol. Phys., 1987, 60, 193 CrossRef.
- J. H. Weiner, John Wiley, New York, Statistical Mechanics of Elasticity , 1983, ISBN 13: 9780471097730 Search PubMed.
- P. E. Werner, L. Eriksson and M. Westdahl, TREOR, a semiexhaustive trial and error powder indexing program for all symmetries, J. Appl. Crystallogr., 1968, 1, 108–113 CrossRef.
- M. Meunier, J. Chem. Phys., 2005, 123, 134906–134907 CrossRef CAS PubMed.
- R. Mills, J. Chem. Phys., 1973, 77, 685–688 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.