
Total syntheses of rubiginone A2, C2, and fujianmycin A†
Abstract
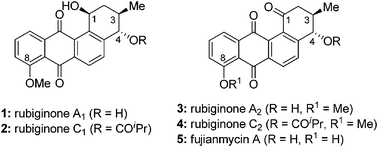
The total syntheses of rubiginone A2, C2 and fujianmycin A are described. The synthesis involves Diels–Alder/aromatization and photo chemical reactions as key steps to construct the tetracyclic frame of benz[a]anthraquinone skeleton. Sharpless epoxidation, a copper-catalyzed regioselective epoxide opening, and enyne metathesis reactions are utilized as the key steps for the synthesis of chiral vinylcyclohexene.
 Please wait while we load your content...
Please wait while we load your content...

