Carbon dioxide adsorption on doped boron nitride nanotubes
Edson N. C. Pauraa,
Wiliam F. da Cunhaa,
João Batista Lopes Martinsb,
Geraldo Magela e Silvaa,
Luiz F. Roncarattia and
Ricardo Gargano*a
aInstitute of Physics, University of Brasilia, Brasília, Brazil. E-mail: gargano@fis.unb.br
bInstitute of Chemistry, University of Brasilia, Brasília, Brazil
First published on 23rd May 2014
Abstract
Boron nitride (BN) nanotubes are promising structures as far as the gas adsorption process is concerned. The electronic and vibrational properties of pristine and cobalt doped single walled boron nitride nanotubes of different chiralities interacting with a carbon dioxide molecule are investigated through the use of density functional theory (DFT) and the discrete variable representation method. When compared to similar simulations concerning carbon nanotubes, a stronger interaction is observed between the carbon dioxide molecule and the functionalized BN nanotube. A density of state investigation suggests that the doping induces major changes in the electronic structure pattern in the sense of critically reducing the original gap. From the vibrational point of view, we note that the zig-zag chirality tends to present higher values of vibrational frequencies for most of the states considered, regardless of the nanotubes being doped or not. Our results suggest that doped zig-zag BN nanotubes are among the best possible candidates for adsorption purposes.
I. Introduction
Due to the current dependence of the global community on fossil fuels as a source of energy, a constant rise in the carbon dioxide (CO2) levels measured in the atmosphere is observed. This behavior has greatly contributed to the greenhouse effect, that in turn implies sudden and undesired climate and environmental changes in our planet.1 In order to overcome, or at least to minimize this deleterious effect, an urgent need to develop efficient and low cost carbon sequestration systems is required.Currently, the scientific community agrees that such systems must have, among other properties, a high surface to volume ratio, chemical and mechanic stability, and a high electronic conductivity, as well as a good catalytic performance. Taking these properties into account, carbon nanotubes (CNTs) stand out as natural gas sensors candidates,2 to be then used as carbon sequestration systems. In this sense, some success has been obtained in detecting and arresting several molecular species (CO2 included) through the use of this kind of system.2–9 As a matter of fact, we have recently performed a thorough investigation of the CO2 adsorption mechanism on cobalt functionalized CNTs.10 It is shown that, from the theoretical point of view, functionalized CNTs are indeed good candidates to be used as CO2 gas sensors, but we also noticed that their sensibility depends on the chirality and the diameter of the system. This feature agrees with the experimental observation that CNTs synthesized by different means may present completely different electronic properties depending on the geometrical characteristics.11 Since controlling these properties is still a challenge for industry, obtaining suitable CNTs to be used as carbon arresting systems on a large scale is currently an unresolved issue. As an alternative route, boron nitride nanotubes (BNNTs) have recently received great attention from the scientific community. As a nanostructure representative, BNNTs also presents a good surface to volume ratio, but unlike CNTs, they tend to exhibit uniform electronic properties regardless of the diameter, chirality and number of layers considered. Add that to the high oxidation and thermal resistances observed,12 greater chemical stability when compared to CNT,13 and fair temperature conductivity14 and one has an interesting class of system for nanotechnology applications, particularly in harmful and oxidative environments in which a CNT would not be so efficient.4 Despite all these favorable properties presented, several studies have established that the doping of BNNTs with transition metals further improves their performance for detecting and adsorbing molecular species. Recent works have shown that the ability of gallium and aluminum doped BNNTs to chemically adsorb the CO, NH3 and SCN molecules is greatly increased in relation to that of the pristine nanotube.15–17 In the same fashion, another study proposed that platinum doped BNNTs might increase the capacity for adsorbing hydrogen molecules.18 As far as the CO2 adsorption in BNNTs is concerned, a recent theoretical investigation conducted by Mousavi and co-workers has proposed that, depending on the adsorption site, the system would turn into either a donor or an acceptor type semiconductor with a reduced gap.19 This study applied a random tight-binding Hamiltonian model, together with the Green’s function methodology to perform a systematic investigation of the adsorption effects of a CO2 specie over the density of states of a zig-zag BNNT (9,0). Their results are expected to be valid for other systems. Concerning the effects of functionalization, it has been reported that by doping a BNNT with boron atoms,20 a chemisorption energy higher than that of that free molecules at room temperature is achieved. According to this work, it is thus, theoretically possible to arrest CO2 at ambient conditions. Similarly, Sun et al. studied the CO2 adsorption on platinum doped armchair BNNT.32 Regardless of whether the substitution site for the platinum atom was nitrogen or boron, once again a reduction of the band gap was observed, together with a considerable charge transfer between the doped BNNT and the CO2 molecule.
Considering those works as a background, one can see that the CO2 adsorption on BNNTs is not fully described, and thus a complete understanding on the influence of many properties over the adsorption mechanism is desired. Therefore, the motivation of this study was due to the fact that there is little information in the literature regarding the effects of chirality in the CO2 adsorption on functionalized BN nanotubes. It was our goal to perform a detailed study, not only on the effects of functionalization over the carbon dioxide adsorption, but also on the role played by chirality and the influence of the active site of adsorption. In order to do so, we performed a systematic investigation on the electronic structure (in the scope of the density functional theory—DFT—within the generalized gradient approximation, GGA) and on the rovibrational spectra (solving the nuclear Schrödinger equation by means of the discrete variable representation) of pristine and doped BNNTs interacting with a CO2 molecule in different sites. Our goal was to investigate how the adsorption mechanism differs from that of CNNTs previously studied. We also sought to investigate whether there is a difference in the adsorption properties depending on the considered active site.
This work is organized as follows: in Section II we describe the main computational features together with the methods used; Section III presents the main results and the description of our calculations; we outline the conclusions in Section IV.
II. Methodology
Our density functional theory calculations consisted of CO2 adsorption on boron nitride nanotubes of an infinite length on a 1D periodic box using the DMol3 code.21,22 The goal was to compute equilibrium geometries, total energies, charge and an electronic density analysis for these complex systems. An electronic exchange correlation was treated using a generalized gradient approximation (GGA)23 with the PW91 functional.24 The positions of all the atoms were fully relaxed until the following convergence criterions were achieved: 10−6 Ha for the total energy, 0.001 Ha Å−1 for the force and 0.003 Å for the displacement. The electronic wave functions were expanded in a 4.4 version double numerical plus polarization basis set (DNP) truncated at a real space cut-off of 4.1 Å.Due to the presence of boron and nitrogen atoms in the models, all the calculations were spin unrestricted. A 0.005 Ha smearing25 and 6 Pulay direct inversion of the iterative subspace (DIIS)26 was applied to the system to ease the convergence of the electronic structures. 3D periodic boundary conditions were applied to the whole system in order to simulate infinitely large BNNTs. The size of the vacuum space inbetween two adjacent tubes was set to be 20 Å, to prevent the interaction of atoms with their periodic images. The Brillouin zone for a single cell was sampled by 1 × 1 × 11 special k-points.
In order to compute the adsorption energy of the CO2 molecules onto the BNNTs from the quantities obtained through our simulations, we adopted the following expression:
| Eb = EBNNT+CO2 − EBNNT − ECO2, | (1) |
In this work, we performed several single point DFT calculations for varying distances between the CO2 molecule and each considered BNNT. We thus obtained a set of points consisting of our potential energy curve—PEC—that we fitted in an analytical expression through the use of a extended Rydberg28 function of the type:
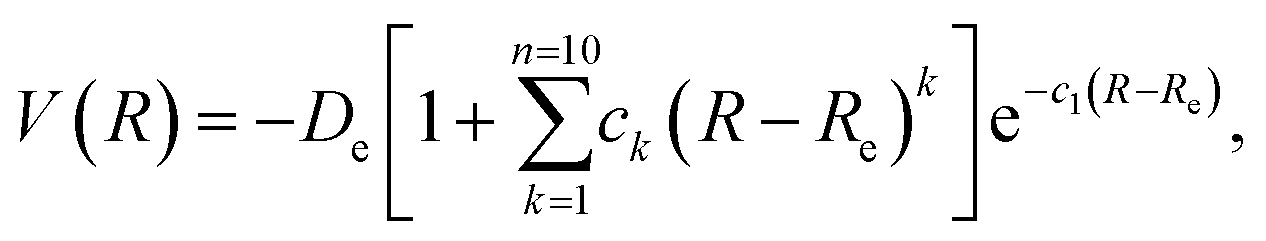 | (2) |
By fitting our electronic energy results with extended Rydberg functions (eqn (2)), we were able to obtain novel analytical expressions for the several considered systems to be used in the solution of Schrödinger nuclear equation through the application of the DVR methodology30 and, in this fashion, to obtain rovibrational energies ε(υ, J), where υ and J respectively denote the vibrational and the rotational quantum numbers. From these energies one can obtain rovibrational spectroscopic constants using the following equations31
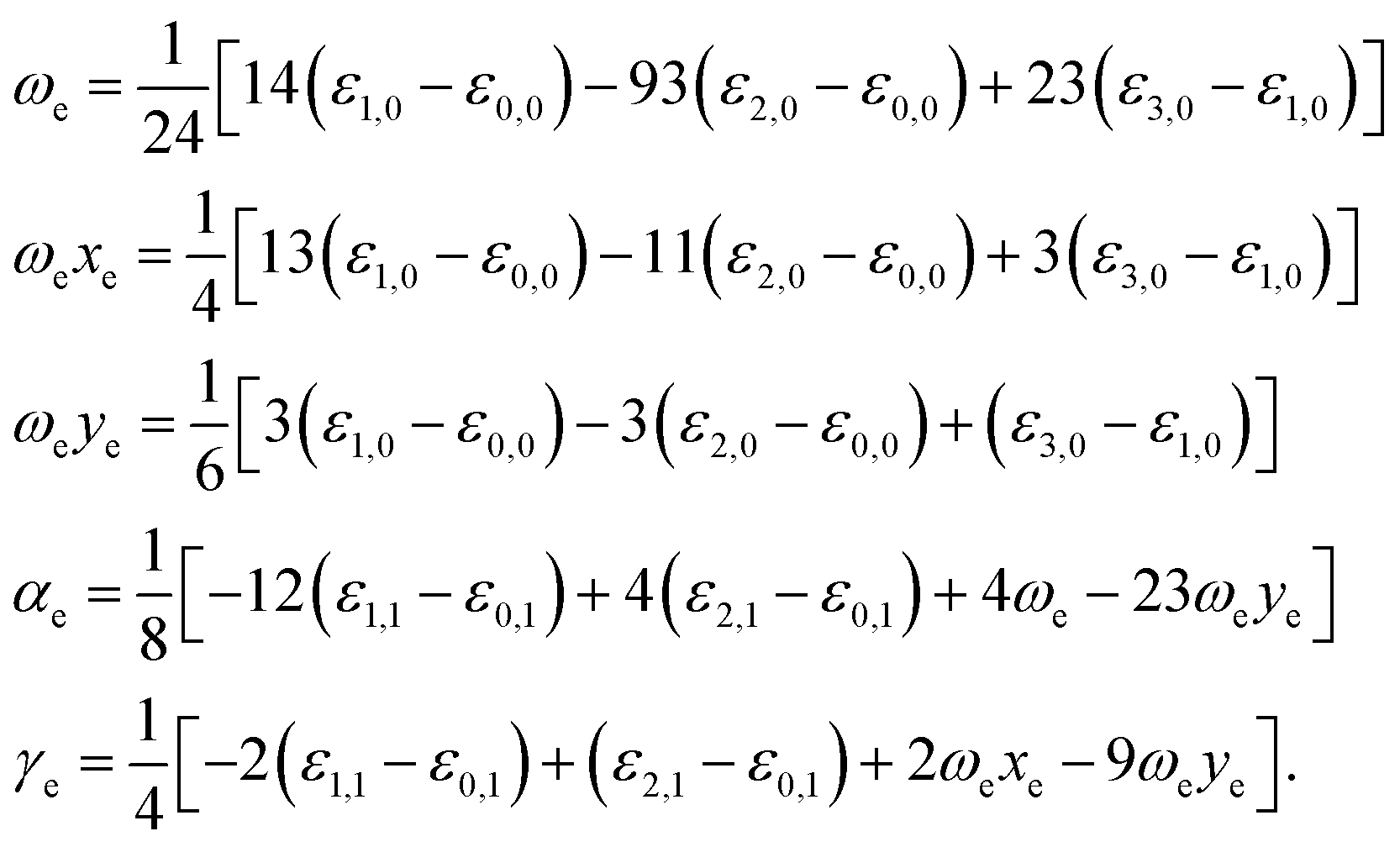 | (3) |
III. Results and discussion
In this work, we investigated the interaction process between pristine and functionalized BNNTs and a carbon dioxide molecule. We used single walled armchair (5,5) and zig-zag (10,0) nanotubes to carry out a comparison between the effects of chirality over the adsorption mechanism. Also, we investigated properties such as the adsorption energy and rovibrational spectra, between the gas molecule and the pristine or cobalt doped nanotube. The goal was to provide an understanding of how the functionalization affects the interaction process.In order to better present our results, we organized the present section into three subsections, each one dealing with one set of simulations. Subsection IIIA regards the adsorption mechanism of the CO2 molecule in pristine BNNTs. In subsection IIIB we investigate the effect of the functionalization of the BNNTs with cobalt. Finally, subsection IIIC is devoted to investigate the adsorption of CO2 molecules in functionalized BNNTs.
A. Pristine BNNTs interacting with a CO2 molecule
In this subsection we discuss the adsorption phenomenon of CO2 molecule in pristine BNNTs of different chiralities, namely armchair (5,5) and zig-zag (10,0). The first step on accomplishing this task was to perform the optimization of the CO2 molecule separately from that of the BNNT (5,5) with its 60 atoms and of BNNT (10,0) with its 80 atoms.The geometry optimization yielded a C⋯O bonding length of 1.166 Å for the carbon dioxide molecule, which is in good accordance with previous results.32 Similarly, geometric results concerning solely the nanotubes reached an excellent agreement with previously published results.33,34 By using the Mulliken method, the charge analysis performed on the armchair nanotube indicated that roughly 0.61e of charge was transferred from the boron atom in the nanotube to the nearby nitrogen sites. As for the zig-zag system, this ratio was observed to be of smaller magnitude as 0.54e of charge was transferred between the B and the N centers. This fact emphasizes the quasi-ionic nature of the B–N bonding in BNNTs. From the density of states (DOS) point of view, together with the band structure analysis, we observed an indirect gap of 4.45 eV for the (5,5) system and a 4.03 eV direct gap for the (10,0), values that present good agreement with data from the literature.33,34
The next step on tackling the adsorption mechanism was to perform the geometry optimization of the complex system composed of CO2 molecule together with the nanotubes. Fig. 1 presents frontal and side view for the equilibrium configuration of the two different systems studied in this work, namely CO2 + BNNT (5,5) (Fig. 1a and b)and O2 + BNNT (10,0) (Fig. 1c and d). It is important to note that, for both cases, neither the CO2 nor the nanotube present structural/geometrical variations when compared to their original equilibrium configuration reached when the optimization was performed separately.
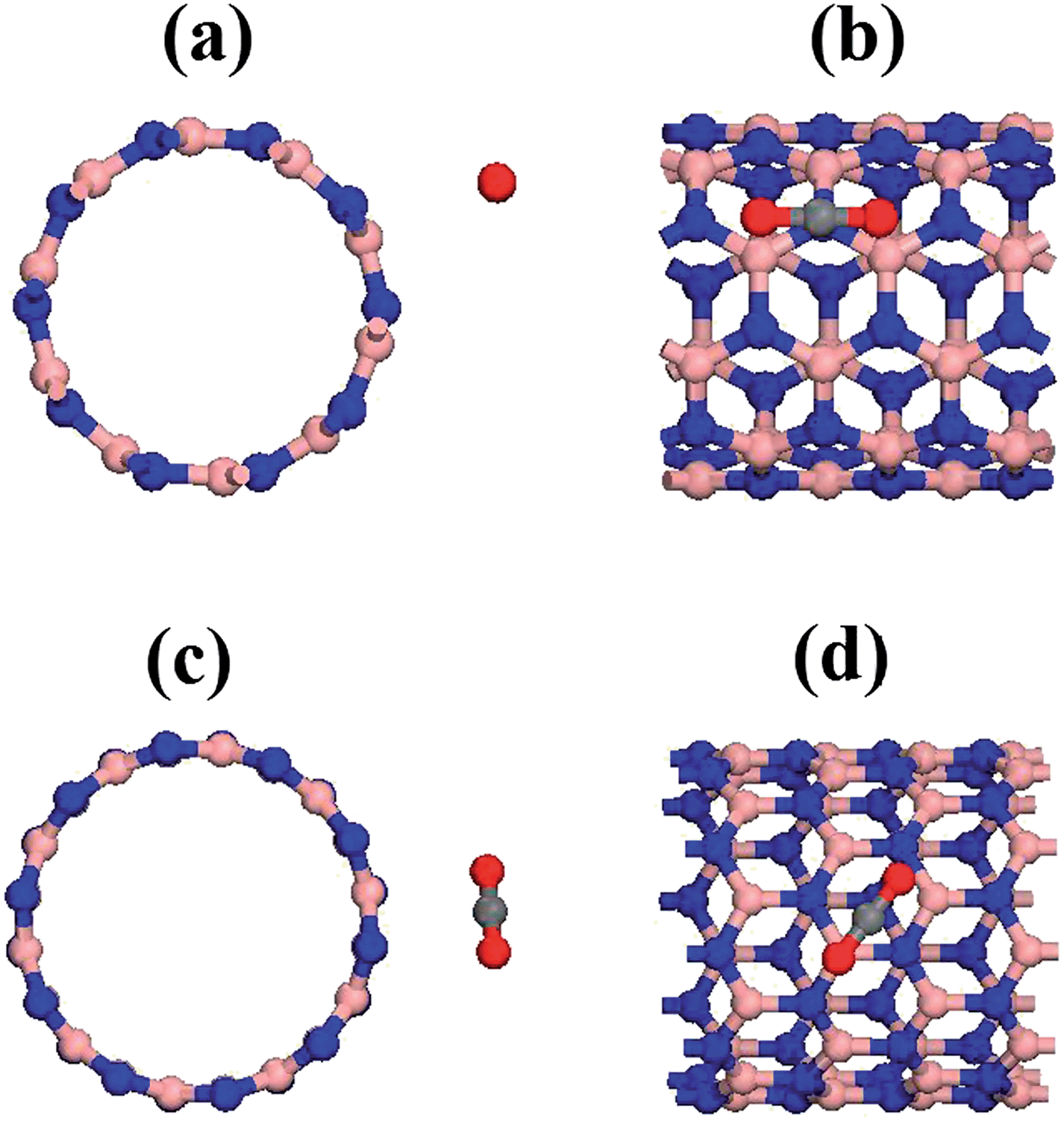 | ||
| Fig. 1 Frontal and side view of the equilibrium distance for the BNNT–CO2 complexes. (a) and (b) armchair BNNT(5,5). (c) and (d) zig-zag BNNT(10,0). | ||
From Fig. 1 the most energetically favorable relative configuration of CO2 + BNNT (5,5) system is the gas molecule aligned parallel to the nanotube axes with the carbon atom of the CO2 molecule standing directly over the nitrogen atom of the nanotube. This configuration can be explained in terms of the polar character of the B–N bonding, that results in a higher electron concentration over the nitrogen atoms when compared to boron. Naturally, these facts directly follow from electronegativity considerations between the atomic species. Thus, one might expect the carbon atom from the gas molecule that, incidentally, presents low electron concentration, to approach the nitrogen center. In the case of the CO2 + BNNT (10,0) system, the configuration is with the carbon atom of CO2 directly over the center of the hexagon in the nanotube wall. The CO2 molecule has a rotation of 38° from its original orientation around its symmetry axis. After this rotation one of the oxygen atoms stands right above a boron atom. In this case, the curvature of the B–N binding in the zig-zag BNNTs plays an important role in the orientation of the molecule relative to the axis of the nanotube, as observed in previous work.35 Taking the optimized geometry of complex systems, the PEC calculations were carried out. The procedure was to systematically approach the CO2 molecule to the nanotube and to compute the corresponding energies, as well as other properties, but always maintaining the subsystems (CO2 and nanotube) geometrically frozen. The main purpose of this work was to analyze the nanotube–CO2 interaction by applying eqn (1) to obtain the adsorption energy. Table 1 presents the equilibrium distances between the closest atom of the BNNTs and carbon of CO2 (D), adsorption energies (Eads), energy gap (Eg) and charge transfer ratio (QT) for the BNNT–CO2 complex system in the two different chiralities considered. One can readily see that the optimized configurations present small binding energies, indicating a poor process of CO2 adsorption in BNNTs, that can be characterized as a physical adsorption that takes place through the mediation of van der Waals (vdW) type interactions. It is important to remark that the results obtained in this subsection are indeed in good agreement with the well known fact that BNNTs are structures with a high level of stability, thus being chemically inert in the absence of dopants.10
| System | Eads (eV) | Eads,vdW (eV) | Eg (eV) | D (Å) | QT (e) |
|---|---|---|---|---|---|
| BNNT (5,5)–CO2 | 0.17 | 0.37 | 4.45 | 3.00 | −0.003 |
| BNNT (10,0)–CO2 | 0.18 | 0.42 | 4.03 | 2.95 | −0.005 |
The comparison between the values obtained for Eads and QT between the armchair and zig-zag BNNTs present a subtle difference of, respectively 0.02 eV and 0.002 e, favoring the former. From Table 1, no difference between the gap energy is achieved when solely considering the adsorption mechanism, which indicates that the presence of the adsorbed molecule does not affect the electronic properties of pristine BNNTs.
B. Functionalizing BNNTs with cobalt
We have seen that the interaction between the carbon dioxide molecule and the pristine BNNTs is of a very weak nature, corresponding to a physical adsorption. Since our goal is to provide a good candidate to carbon arresting system, this interaction must be strengthened. In order to do so, we functionalized the nanotube through a substitutional doping with a cobalt atom. Because of the very nature of BNNTs, one should expect a different behavior depending on which is the substituted atom on the nanotube lattice, i.e., boron or nitrogen.Considering n and m, the indexes that take chirality into account, when a cobalt atom substitutes a boron atom, we used the notation BNNT(n,m)CoB. Alternatively, when the substituted atom is nitrogen, we used the notation BNNT(n,m)CoN. A new complete geometry optimization was performed for the system composed of BNNTs with the substitutional Co atom. The optimized geometries for doped BNNTs are presented in Fig. 2, in which the geometric configuration for the cobalt atom related to the adjacent boron and nitrogen species are highlighted for both chiralities. One can readily see that the geometrical structure of the doped BNNTs is modified. Particularly, the cobalt atom is projected outside the nanotubes wall, due to its greater size related to the rest of the chain.
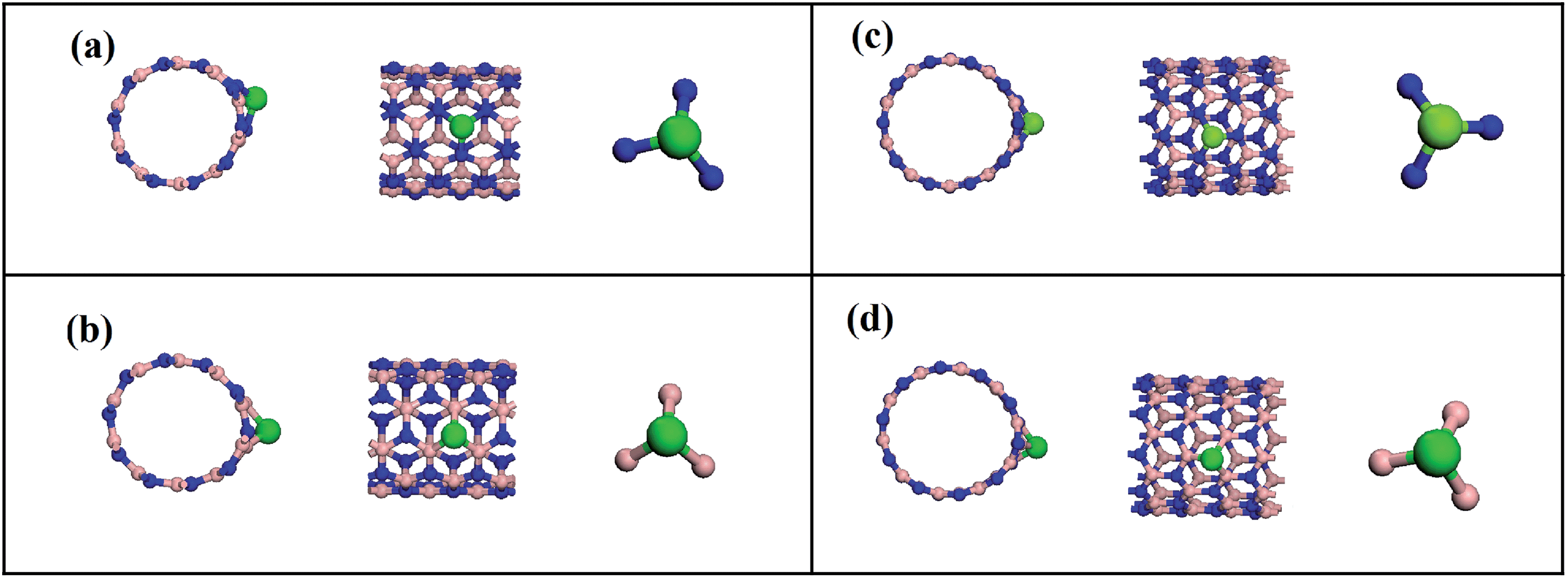 | ||
| Fig. 2 Frontal and side view of the optimized geometries for cobalt doped BNNTs (a) BNNT(5,5)CoB, (b) BNNT(5,5)CoN, (c) BNNT(10,0)CoB, (d) BNNT(10,0)CoN. Geometric configuration for the cobalt atom is highlighted. | ||
One can see that for both BNNT (5,5) and (10,0), substituting a nitrogen atom causes a larger geometric deformation on the nanotubes in relation to the boron substitution. This fact is confirmed by noting that the average length of the Co–N bond is around 1.70 Å, which is smaller than the Co–B length of roughly 1.90 Å. Therefore, substituting a boron atom yields a smaller difference on the binding length related to the typical B–N of 1.45 Å for the pristine nanotube. It is important to note that these geometrical deformations lead to important changes in the electronic properties for the nanotubes.
Fig. 3 shows the density of states (DOS) calculated for the nanotubes that show how the addition of a cobalt atom affects the electronic distribution of the BNNTs. Adding a cobalt atom to the BNNTs induces the appearance of some impurity states near the Fermi level, thus resulting in a gap decrease (Fig. 3). Particularly, for the BNNT(5,5)CoB the appearance of a state at the end of the valence band reduces the original gap to 2.57 eV. In the case of BNNT(5,5)CoN we should stress the appearance of three states, two of those on the end of the valence band and the other on top of the conduction band. In this case, the energy gap is even further decreased to 1.63 eV. BNNT(10,0)CoB shows a similar pattern to BNNT(5,5)CoB, thus presenting a final gap of 2.13 eV. However, for the zig-zag nanotube it is observed an electronic state much closer to the valence band. BNNT(10,0)CoN, on the other hand, presents a whole different behavior when it comes to the functionalization induced change of density of states. In this case, states next to the conduction band together with a gap reduction to 2.13 eV were obtained. The partial DOS reveals that these impurities states are mainly due to the electrons of the cobalt atom and the π electrons of the three cobalt neighbors in the BNNTs.
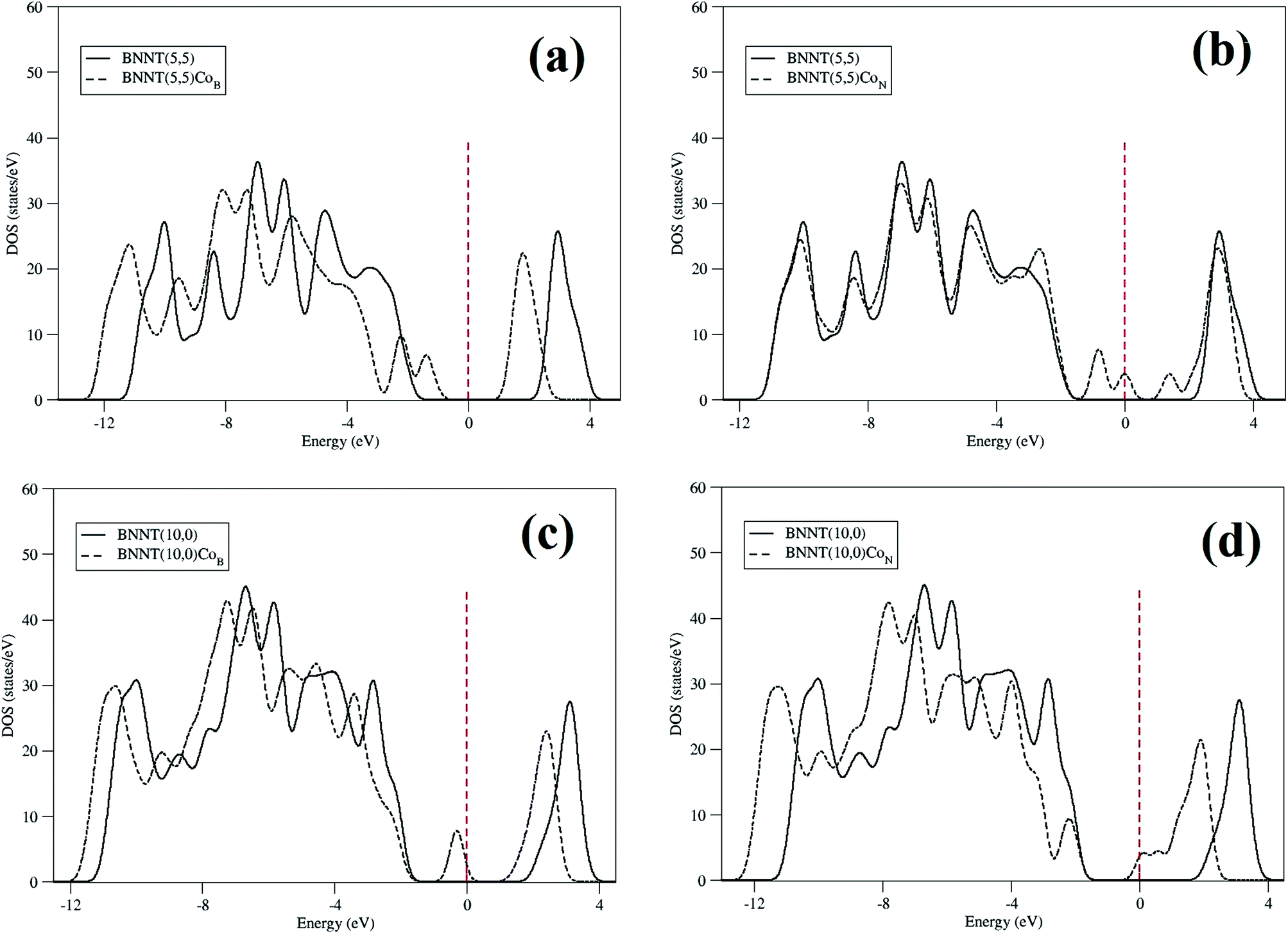 | ||
| Fig. 3 DOS for pristine (solid line) and doped (dashed line) BNNTs. (a) BNNT(5,5)CoB, (b) BNNT(5,5)CoN, (c) BNNT(10,0)CoB, (d) BNNT(10,0)CoN. | ||
The Mulliken charge analysis indicates that a certain amount of electrons is transferred from the cobalt center to the BNNT. It is important to note that, for both chiralities, the amount of charge transferred in the case of BNNT(n,m)CoN is smaller than in the case of BNNT(n,m)CoB. This can be explained by remembering that the electronegativity of nitrogen is larger than that of boron, therefore leading the former to attract cobalt.
C. Cobalt doped BNNTs interacting with CO2 molecule
The equilibrium configurations for the optimized geometries of different simulations are depicted in Fig. 4. We have found a difference compared to the non-doped structure: the optimized geometry yields a symmetry breaking of the CO2 molecule for all the cases, i.e., the gas molecule changes from a linear to a bent configuration. This trend is related to a chemical adsorption process because the bent structure is expected in the case of highest binding energy.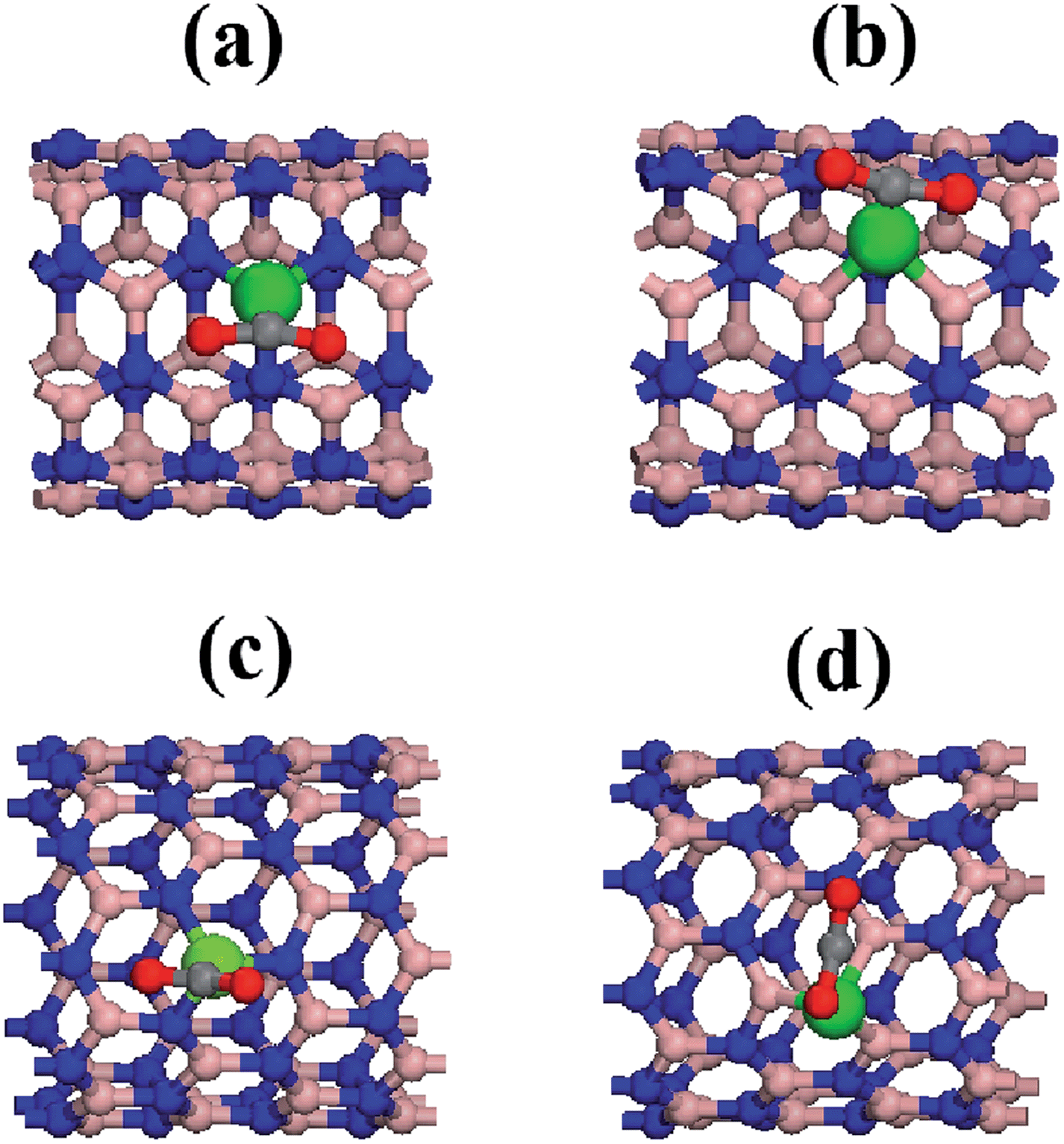 | ||
| Fig. 4 Frontal and side view of the equilibrium configuration of the CO2 related to cobalt doped BN nanotubes (a) BNNT(5,5)CoB, (b) BNNT(5,5)CoN, (c) BNNT(10,0)CoB, (d) BNNT(10,0)CoN. | ||
Table 2 presents a comparison between Eads, D, Eg and QT values for the cobalt doped BNNTs interacting with CO2. The first interesting feature to be noted is that the presence of cobalt atoms significantly raises the adsorption energy for all complexes.
| System | Eads (eV) | Eads,vdW (eV) | Eg (eV) | D (Å) | QT (e) |
|---|---|---|---|---|---|
| BNNT(5,5)CoB–CO2 | 0.61 | 0.79 | 1.63 | 2.00 | −0.086 |
| BNNT(5,5)CoN–CO2 | 1.02 | 1.12 | 0.89 | 1.90 | −0.180 |
| BNNT(10,0)CoB–CO2 | 0.67 | 0.87 | 1.50 | 2.10 | −0.102 |
| BNNT(10,0)CoN–CO2 | 2.54 | 2.79 | 1.00 | 1.95 | −0.247 |
Either for the BNNT(5,5) or for BNNT(10,0) the substitution of a nitrogen for a cobalt atom yields larger values for the adsorption energy than the correspondent substitution of a boron atom. The 1.02 eV energy obtained for BNNT(5,5)CoN–CO2 is 0.86 eV larger than the BNNT(5,5)–CO2 case, indicating that the interaction of the carbon dioxide molecule with this functionalized armchair tube is far superior to that of the pristine system. The equilibrium distance between the carbon and cobalt atom was observed to be of 1.90 Å, which, together with the energy values, suggests a strong chemical interaction between the species. Table 2 also shows that the charge transfer between BNNT(5,5)CoN to the CO2 molecule is of 0.156 e and that the energy gap is reduced from 1.63 eV to 0.87 eV, indicating that the electronic properties of free BNNT(5,5)CoN are drastically changed after the adsorption process takes place. The BNNT(5,5)CoB–CO2 configuration presented an interaction distance of 2.0 Å and a charge transfer ratio of 0.057, thus consisting in a much poorer candidate to adsorbing system. Note that this configuration yielded smaller values of adsorption energy than the previous case of BNNT(5,5)CoN.
An even more interesting result is observed when considering the simulations for BNNT (10,0). Considering the functionalized BNNT(10,0)CoN both the carbon and the oxygen atoms of the molecule simultaneously interact with the cobalt atom in the nanotube. The C–Co distance was of around 1.95 Å whereas the O–Co was 1.87 Å. The obtained adsorption energy was of 2.54 eV, which is 1.52 eV larger than the one for BNNT(5,5)CoN–CO2, indicating that for this dopant, the zig-zag chirality is more efficient than the armchair in the capture of CO2 molecule. It is important to note that these values for the adsorbing energy are far superior to those for a single walled carbon nanotube similarly doped.15 This remarkable result indicates that BN nanotubes are better candidates for CO2 arresting when compared to carbon nanotubes, which gives 0.12 eV of adsorption energy.10 The charge transfer between the molecule and the nanotube was of 0.247 e, demonstrating the chemical bonding character obtained in these cases. Due to the reduction from 4.03 eV to 1.00 eV of the gap, we can see that substituting nitrogen by cobalt provides the nanotube with electronic transport properties that were absent in the previous case. For the case of BNNT(10,0)CoB–CO2 a 0.67 eV value was achieved for the adsorption energy, and 0.065e for the charge transfer. These values are superior to those presented for the BNNT(5,5)CoB–CO2 system, which confirms our previous statement about the zig-zag chirality being more effective than the armchair in the present case. These results are in accordance with the recent study of carbon nanotubes showing that the zigzag chirality present high adsorption energies than the armchair.10
In order to fully understand the electronic properties of the complex systems composed of functionalized BNNTs interacting with the carbon dioxide molecule, we made use of the total charge density and the density of electronic states. Fig. 5 presents an isosurface calculation for the total density of the most stable geometrical configuration of each chirality. As shown, for the pristine BNNTs, the electronic density of the tube was isolated to that of the molecule. In this case, no considerable charge overlap is perceived, thus confirming the weak interaction between these species. This situation is in striking contrast to the one observed when the tubes are functionalized. In this case, a considerable overlap of the electronic density between the nanotube and the molecule is observed, indicating a strong interaction between the correspondent orbitals and a clear electronic flux from the tube to the molecule. Focusing on BNNT(5,5)CoB, the charge superposition involves only the carbon atom and presents itself as a carbon coordination.36 In the case of BNNT(10,0)CoN, the charge overlap also involves the oxygen atom, thus consisting on a side-on coordination. We believe that the coordination of CO2 on Co is stabilized mainly due to electrostatic interactions and back-bonding process, in the same fashion as reported by Morokuma et al. for N2 complexes.36
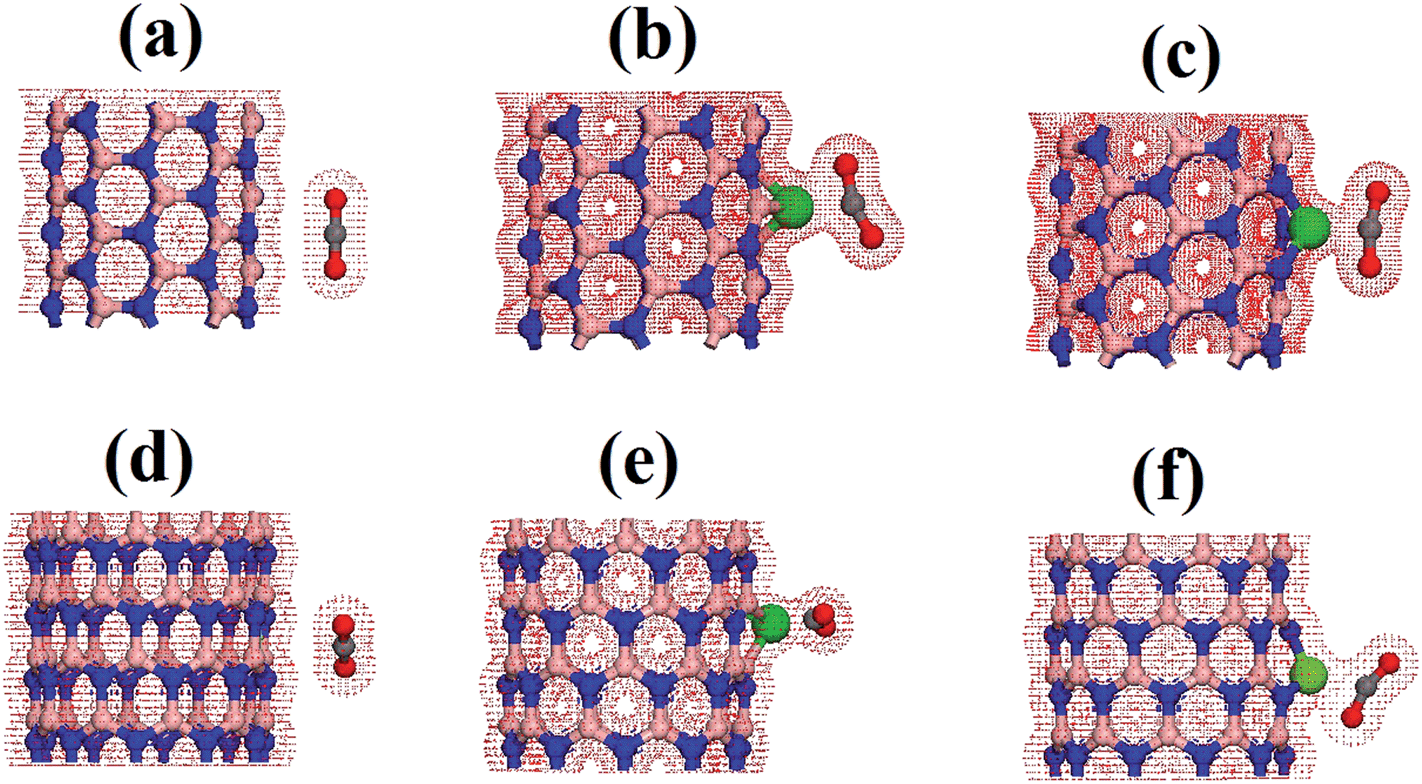 | ||
| Fig. 5 Charge density for pristine and cobalt doped BNNT interacting with the CO2 (a) BNNT(5,5), (b) BNNT(5,5)CoN, (c) BNNT(5,5)CoB, (d) BNNT(10,0), (e) BNNT(10,0)CoN, (f) BNNT(10,0)CoB. | ||
We present, in Fig. 6, the DOS comparison for all the configurations considered in this work. The DOS for pristine BNNTs interacting with the CO2 molecule did not present any considerable change when compared to the case in which the nanotubes are isolated (Fig. 3). This means that the electronic properties of BNNTs are not influenced by the presence of the CO2 molecule, which is another way to state the poor interaction between the species in this case. BNNT(5,5)CoN–CO2 shows an electronic state at the end of the valence band along with another one on the top of the conduction band. For the BNNT(10,0)CoN–CO2, the same tendency was observed. Fig. 6 shows a decrease of the DOS intensity after CO2 interaction, compared to Fig. 3 without CO2, for all cases. Furthermore, we found a shift of the bands near the Fermi level towards lower energies. This behavior is probably due to the interaction of empty Co ”d” originated from the cobalt atom and of the π electrons from the CO2 molecule in the gap region of the nanotubes. This new configuration positively affects the chemisorption capacity of the nanotubes, thus improving their reactiveness with the gas molecule.
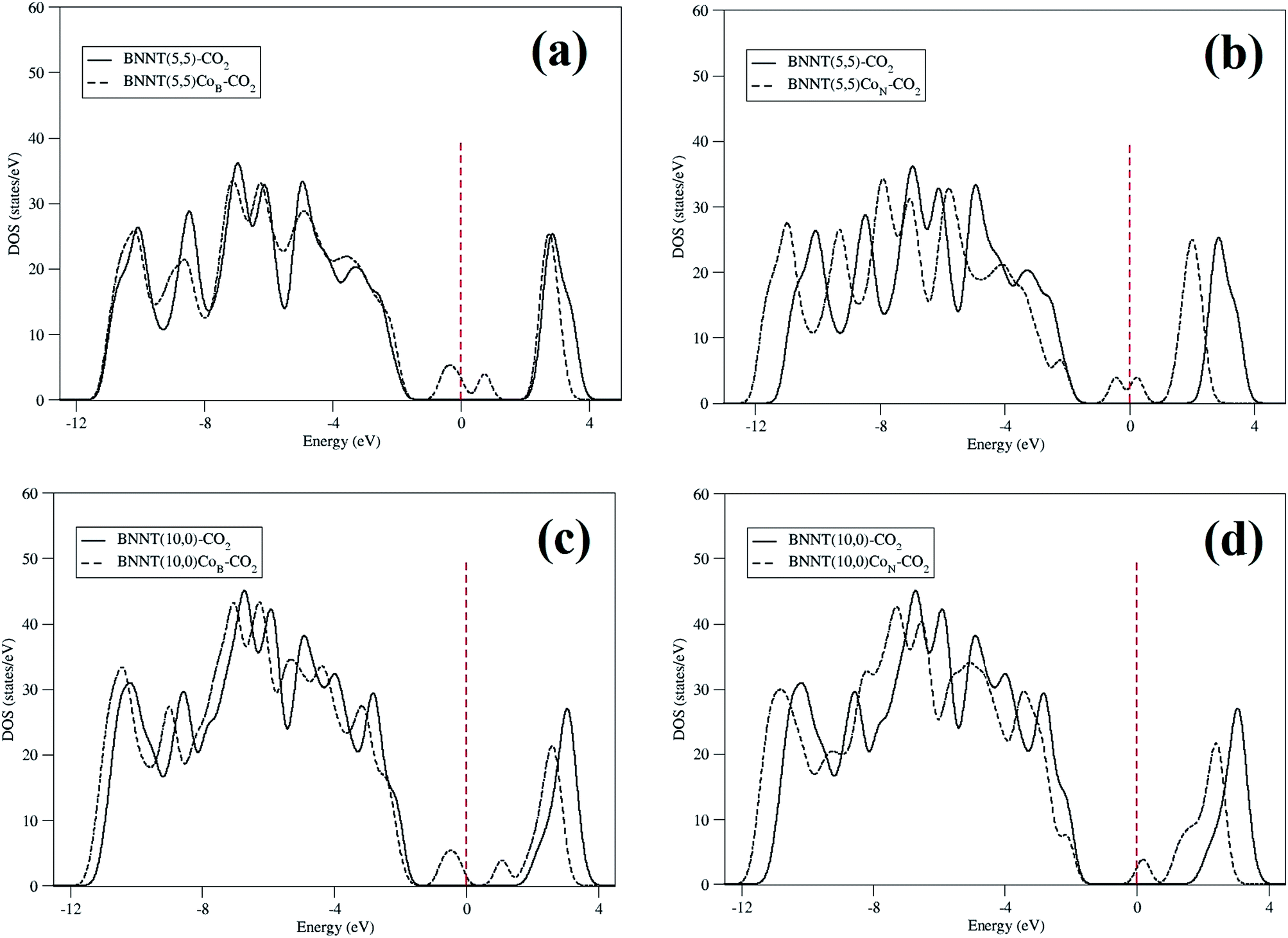 | ||
| Fig. 6 DOS comparison for pristine (solid line) and doped (dashed line) BNNTs interacting with CO2. (a) BNNT(5,5)CoB, (b) BNNT(5,5)CoN, (c) BNNT(10,0)CoB, (d) BNNT(10,0)CoN. | ||
An interesting feature of the DOS plots is the Fermi level shifting towards lower energies caused by the Co doping found in the BNNT (5,5). This shifting is a direct consequence of cobalt substitution in the nanotube. This is probably due to the charge transfer from the cobalt atom to the neighboring atoms of the nanotube. However, this shift occurs differently for both substitutions: 0.90 eV and 0.35 eV for BNNT(5,5)CoB and BNNT(5,5)CoN, respectively. In this case a charge transfer of 0.016 a.u. for BNNT(5,5)CoB and 0.019 a.u for BNNT(5,5)CoN was observed. The same trend was also verified in the BNNT(10,0)CoB and BNNT(10,0)CoN, in which the values were of 1.02 eV and 0.40 eV, respectively. A charge transfer of 0.40 a.u. for BNNT(10,0)CoB and 0.34 a.u for BNNT(10,0)CoN took place. For the nanotubes interacting with CO2 molecule, the Fermi level was shifted towards lower energies, considering that there is a charge transfer from the nanotube to the CO2 molecule. The shift was of 0.38 eV for BNNT(5,5)CoB, 0.66 eV for BNNT(5,5)CoN, 0.39 eV for BNNT(10,0)CoB and 0.90 eV for BNNT(10,0)CoN.
Fig. 7 presents the adsorption energy curve as a function of the equilibrium distance for the complex BNNT(n,m)Co–CO2. Through all the cases, the curves were constructed taking the cobalt to carbon distance into account. The considered distances were taken from the strong repulsion to the asymptotic region of the potential. It is important to note that the PEC was carried out using frozen structures. These curves were used in determining the vibrational spectrum of all the considered tubes. We should also remark that the high energy of the wall obtained for BNNT(10,0) + CoN–CO2, also demonstrates that this is a system with great potential of applicability in a chemisorption process.
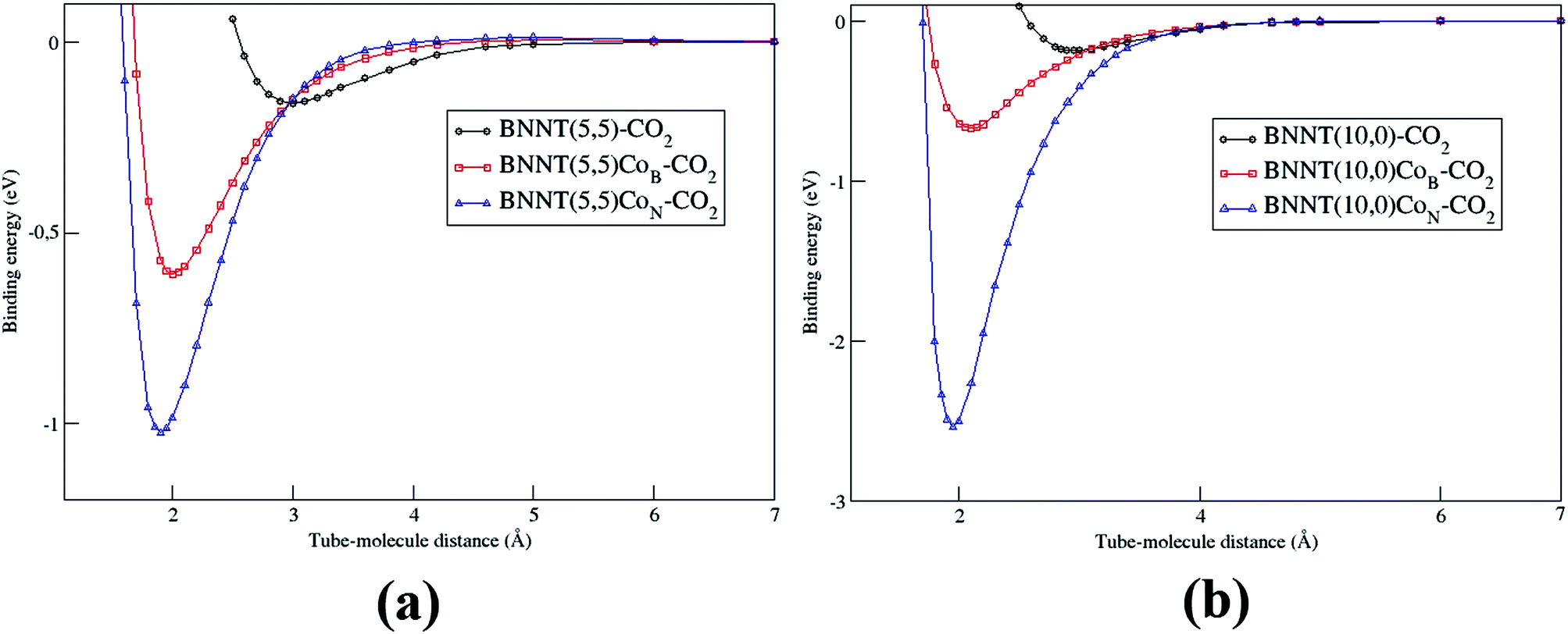 | ||
| Fig. 7 Comparison between the PECs for pristines and doped BNNTs interacting with the CO2 molecule. (a) BNNT (5,5) e (b) BNNT(10,0). | ||
Van der Waals corrections were carried out for all the studied systems. We found a contribution of the van der Waals correction (Table 1) for the physical adsorption. On the other hand, chemical adsorption energies show a small contribution of the vdW correction (Table 2). Furthermore, the difference of pristine and doped BNNTs adsorption energies, showed the same trend with or without vdW correction (Table 3).
| System | ΔEads (eV) | ΔEads,vdW (eV) |
|---|---|---|
| BNNT(5,5)CoB–CO2 | 0.42 | 0.44 |
| BNNT(5,5)CoN–CO2 | 0.76 | 0.85 |
| BNNT(10,0)CoB–CO2 | 0.44 | 0.49 |
| BNNT(10,0)CoN–CO2 | 2.37 | 2.36 |
The nuclear Schrödinger equation was solved using the analytical form of the potential obtained using the data in Table 4. Through this formalism we investigate the vibrational spectra of the system and, as a consequence, the spectroscopic constants.
| Coefficients (Å−k) | BNNT(5,5) | BNNT(5,5)CoB | BNNT(5,5)CoN | BNNT(10,0) | BNNT(10,0)CoB | BNNT(10,0)CoN |
|---|---|---|---|---|---|---|
| C1 | 0.4010528 × 101 | 0.5232746 × 101 | 0.4600582 × 101 | 0.4567201 × 101 | 0.6115366 × 101 | 0.7040093 × 101 |
| C2 | 0.5030727 × 101 | 0.9369224 × 101 | 0.5677401 × 101 | 0.7005336 × 101 | 0.1441551 × 102 | 0.1816739 × 102 |
| C3 | 0.1732614 × 101 | 0.1411859 × 102 | 0.6954038 × 101 | 0.1063563 × 102 | 0.1539079 × 102 | 0.2356444 × 102 |
| C4 | 0.3538602 × 101 | 0.1262842 × 102 | 0.6161214 × 101 | 0.1408899 × 102 | 0.1261757 × 102 | 0.2091481 × 102 |
| C5 | 0.1570261 × 102 | −0.1994591 × 102 | −0.1954557 × 102 | −0.1239502 × 102 | 0.5521242 × 102 | 0.1418446 × 103 |
| C6 | −0.6353567 × 101 | 0.1075792 × 102 | −0.1865900 | −0.5775726 × 101 | 0.3535850 × 102 | 0.3083373 × 102 |
| C7 | −0.2287133 × 102 | 0.6586673 × 102 | 0.5043954 × 102 | 0.3746132 × 102 | −0.8246642 × 102 | −0.4739153 × 103 |
| C8 | 0.2267056 × 102 | −0.9270648 × 102 | −0.5766459 × 102 | −0.3842283 × 102 | 0.7531488 × 102 | 0.8008445 × 103 |
| C9 | −0.7336704 × 101 | 0.4957445 × 102 | 0.2504332 × 102 | 0.1522006 × 102 | −0.2135814 × 102 | −0.4811759 × 103 |
| C10 | 0.7945204 | −0.9315078 × 101 | −0.3925971 × 101 | −0.1905156 × 101 | 0.7613046 × 101 | 0.1169292 × 103 |
Table 5 presents the results of the eleven first vibrational states for the pristine and cobalt doped complexes. Higher values of the vibrational energies for the doped systems were found when compared with the pristine BNNTs. We can also see that, typically, substituting the cobalt in the nitrogen site yields an even greater vibrational energy for all the cases when compared to the boron, regardless of the chirality considered. Also, the zig-zag chirality presented greater values of vibrational frequency when compared with the armchair chirality. This result is consistent with this chirality presenting a potential well of greater depth than that of the armchair, as already mentioned. The electron transfer of backbonding strengthens the Co–CO2 bonding and should be related to the increase of this bonding vibration.
| ν | BNNT(5,5)–CO2 | BNNT(5,5)CoB–CO2 | BNNT(5,5)CoN–CO2 | BNNT(10,0)–CO2 | BNNT(10,0)CoB–CO2 | BNNT(10,0)CoN–CO2 |
|---|---|---|---|---|---|---|
| 0 | 39.09 | 92.09 | 127.29 | 43.99 | 95.25 | 231.77 |
| 1 | 115.42 | 272.05 | 377.76 | 127.05 | 283.54 | 692.39 |
| 2 | 188.44 | 447.47 | 623.17 | 204.96 | 467.85 | 1148.42 |
| 3 | 258.23 | 618.96 | 863.90 | 279.28 | 648.51 | 1599.78 |
| 4 | 324.83 | 786.90 | 1100.29 | 350.96 | 825.74 | 2046.32 |
| 5 | 388.36 | 951.54 | 1332.61 | 420.49 | 999.62 | 2487.83 |
| 6 | 449.03 | 1113.02 | 1561.10 | 488.12 | 1170.11 | 2924.08 |
| 7 | 507.15 | 1271.39 | 1785.93 | 553.97 | 1337.08 | 3354.86 |
| 8 | 563.08 | 1426.68 | 2007.23 | 618.07 | 1500.38 | 3779.94 |
| 9 | 617.18 | 1578.86 | 2225.12 | 680.42 | 1659.85 | 4199.10 |
| 10 | 669.71 | 1727.90 | 2439.65 | 741.02 | 1815.35 | 4612.17 |
By taking the differences between the rows of Table 5 for a given column, one can obtain the shift on the spectrum for each system. We thus obtained energy red shifts concerning the transitions from the states listed in Table 5. By carrying out this analysis one can note that the red shifts are accompanied by a shortening of the BNNTCo–CO2 equilibrium distances. These result mean that the pristine BNNT–CO2 intermolecular interaction energies are weaker than that of BNNTCo–CO2, due to the interaction of CO2 molecule with Co and the nanotube. The red shifts obtained belong to the far and mid-infrared portions of the electromagnetic spectrum, as expected for intermolecular interactions.
The first striking evidence to be observed is that the doping strongly affects the shifts in the sense of increasing their energies. This fact is expected from the deepening of the potential well, due to this doping, as previously observed in the PECs analysis. Another clear feature is that the transitions on the zig-zag chirality are more energetic than those of the armchair. This happens to be true when we compare the same kind of functionalization. As far as functionalization is concerned, we can see that, for both chiralities, when the cobalt dopant substitutes the nitrogen atom, a higher energy shift pattern is observed. This fact is consistent with the other results.
Finally, by applying eqn (3), we obtain the spectroscopic values for all the BNNT–CO2 complexes in Table 6. From this table, it is possible to note that the BNNTCo–CO2 vibrational frequency constants, ωe, are greater than ωe for the pristine BNNT–CO2. This feature shows that in the BNNTCo–CO2 complex, the frequencies are more harmonic than for the pristine BNNT–CO2 complex. The BNNT–CO2 and BNNTCo–CO2 intermolecular rotation–vibration interaction constants (αe and γe) are very small, which means the rotational and vibrational modes are little coupled. This is expected as the mass of CO2 is negligible compared to the nanotube. We can also see that when cobalt substitutes for nitrogen in the nanotube, the frequency tends to be enhanced, a fact that, again, is consistent with our other results. These results are of major importance for future spectroscopic works, and may be used for comparison whenever experimental and theoretical studies on carbon dioxide adsorption on carbon nanotubes are considered.
| Spectroscopic constants | BNNT(5,5) | BNNT(5,5)CoB | BNNT(5,5)CoN | BNNT(10,0) | BNNT(10,0)CoB | BNNT(10,0)CoN |
|---|---|---|---|---|---|---|
| ωe (cm−1) | 79.6765 | 185.0764 | 255.9174 | 89.7255 | 192.6107 | 465.111 |
| ωexe (cm−1) | 1.6922 | 2.7222 | 2.8298 | 3.7563 | 2.2535 | 2.2221 |
| ωeye (cm−1) | 0.00965 | 0.10067 | 0.06572 | 0.26174 | 0.05768 | −0.01526 |
| αe (cm−1) | 0.0003 | 0.00163 | 0.0004 | 0.0012 | 0.0002 | 0.0003 |
| γe (cm−1) | −5.2063 × 10−5 | 5.3221 × 10−5 | 1.8543 × 10−5 | 5.6580 × 10−5 | −5.2871 × 10−5 | −2.3347 × 10−5 |
IV. Conclusions
In this work, we carried out DFT ab initio calculations within the GGA to study the interaction of a CO2 molecule with two different sites on the surface of pristine and doped BNNTs. Both the armchair (5,5) and the zig-zag (10,0) chiralities were investigated. We found that the CO2 molecule interacts weakly with pristine BNNTs through van der Waals like interactions. By carrying out the doping with a cobalt atom, a considerable increase on the binding energy between the CO2 and the nanotube was noted. We developed a set of simulations to draw comparisons between the adsorption energy for the cobalt atom substituting either the nitrogen or the boron atom for both nanotubes chiralities. It was concluded that the most suitable system as far as the adsorption energy is concerned is the doped zig-zag system in which the a nitrogen was substituted by the cobalt dopant. We noted that the BNNT(10,0)CoN–CO2 complex presented chemisorption states far superior than those observed for carbon nanotubes, whose data are present in the literature.10 The better quality of BNNTs when compared to carbon nanotubes, as potential candidates as gas adsorbents, were confirmed through DOS investigations, a Mulliken charge analysis and also from the vibrational spectrum. As for this latter analysis, we noted vibrational frequencies of systematically higher values than those for carbon nanotubes. These facts, together with advantages such as a great chemical stability and the ease of synthetic control that BNNTs present, leads us to consider doped BNNTs as promising structures for the detection, capture and arresting of CO2 molecules in different media. Therefore, we have shown that these systems are tools of significant importance for the challenge of developing an environmentally desired mechanism of carbon arresting.Acknowledgements
The authors gratefully acknowledge the financial support from the Brazilian Research Councils CNPq, CAPES, and FINATEC.References
- S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Nature, 2011, 476, 43 CrossRef CAS PubMed.
- J. Kong, N. Franklin, C. Zhou, M. Chapline, S. Peng, K. Cho and H. Dai, Science, 2000, 287, 622 CrossRef CAS.
- P. G. Collins, K. Bradley, M. Ishigami and A. Zettl, Science, 2000, 287, 1801 CrossRef CAS.
- J. Li, Y. J. Lu, Q. Ye, M. Cinke, J. Han and M. Meyyappan, Nano Lett., 2003, 3, 929 CrossRef CAS.
- A. Goldoni, R. Larciprete, L. Petaccia and S. Lizzit, J. Am. Chem. Soc., 2003, 125, 11329 CrossRef CAS PubMed.
- J. Zhao, A. Buldum, J. Han and J. P. Lu, Nanotechnology, 2002, 13, 195 CrossRef CAS.
- M. Cinke, J. Li, C. W. Bauschlicher, A. Ricca and M. Meyyappan, Chem. Phys. Lett., 2003, 376, 761 CrossRef CAS.
- J. M. Garcia-Lastra, D. J. Mowbray, K. S. Thygesen, A. Rubio and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 081408 CrossRef.
- C. Matranga and B. Bockrath, J. Phys. Chem. B, 2004, 108, 6170 CrossRef CAS PubMed.
- E. N. P. Costa, W. F. da Cunha, P. H. de Oliveira Neto, G. M. e Silva, J. B. L. Martins and R. Gargano, J. Phys. Chem. A, 2013, 117, 2854 CrossRef PubMed.
- J. Beheshtian, A. A. Peyghan and Z. Bagheri, Sens. Actuators, B, 2012, 171, 846 CrossRef PubMed.
- A. P. Suryavanshi, M. Yu, J. Wen, C. Tang and Y. Bando, Appl. Phys. Lett., 2004, 84, 2527 CrossRef CAS PubMed.
- E. Bengu and L. D. Marks, Phys. Rev. Lett., 2001, 86, 2385 CrossRef CAS.
- C. Zhi, Y. Bando, C. Tang and D. Golberg, Mater. Sci. Eng., R, 2010, 70, 92 CrossRef PubMed.
- A. A. Peyghan, A. Soltani, A. A. Pahlevani, Y. Kanani and S. Khajeh, Appl. Surf. Sci., 2013, 270, 25 CrossRef CAS PubMed.
- A. Soltani, S. G. Raz, V. J. Rezaei, A. D. Khalaji and M. Savar, Appl. Surf. Sci., 2012, 263, 619 CrossRef CAS PubMed.
- A. Soltani, N. Ahmadian, Y. Kanani, A. Dehnokhalaji and H. Mighani, Appl. Surf. Sci., 2012, 258, 9536 CrossRef CAS PubMed.
- X. Wu, J. L. Yang and X. C. Zenga, J. Chem. Phys., 2006, 125, 044704 CrossRef PubMed.
- H. Mousavi, J. M. Kurdestany and M. Bagheri, Appl. Phys. A: Mater. Sci. Process., 2012, 108, 283 CrossRef CAS.
- H. Choi, Y. C. Park, Y. H. Kim and Y. S. Lee, J. Am. Chem. Soc., 2011, 133, 2084 CrossRef CAS PubMed.
- Discovery Studio Modeling Environment, Release 3.5, San Diego, Accelrys Software Inc., 2012 Search PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter, 1992, 46, 6671 CrossRef CAS.
- M. Weinert and J. W. Davenport, Phys. Rev. B: Condens. Matter, 1992, 45, 13709 CrossRef.
- P. Pulay, J. Comput. Chem., 1982, 3, 556 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- J. N. Murrel, S. Carter, S. C. Farantos, P. Huxley and A. J. C. Varandas, Molecular Potential Energy Functions, Wiley, Chinchester, 1984, pp. 1–11 Search PubMed.
- M. J. D. Powell, Comput. J., 1964, 7, 155 CrossRef.
- J. J. S. Neto and L. S. Costa, Braz. J. Phys., 1998, 28, 1 CrossRef PubMed.
- H. V. R. Vila, L. A. Leal, L. A. Ribeiro, J. B. L. Martins, G. M. e Silva and R. Gargano, J. Mol. Spectrosc., 2012, 273, 26 CrossRef PubMed.
- Q. Dong, X. M. Li, W. Q. Tian, X. R. Huang and C. C. Sun, THEOCHEM, 2010, 948, 83 CrossRef CAS PubMed.
- S. F. Wang, Y. Zhang, J. M. Zhang, K. W. Xu and J. Vincent, Appl. Phys. A: Mater. Sci. Process., 2012, 109, 601 CrossRef CAS PubMed.
- P. Piquini, R. J. Baierle, T. Schmidt and A. Fazzio, Nanotechnology, 2005, 16, 827 CrossRef CAS.
- Y. Li, Z. Zhou and J. Zhao, Nanotechnology, 2008, 19, 015202 CrossRef PubMed.
- S. Sakaki, K. Kitaura and K. Morokuma, Inorg. Chem., 1982, 21, 760 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |