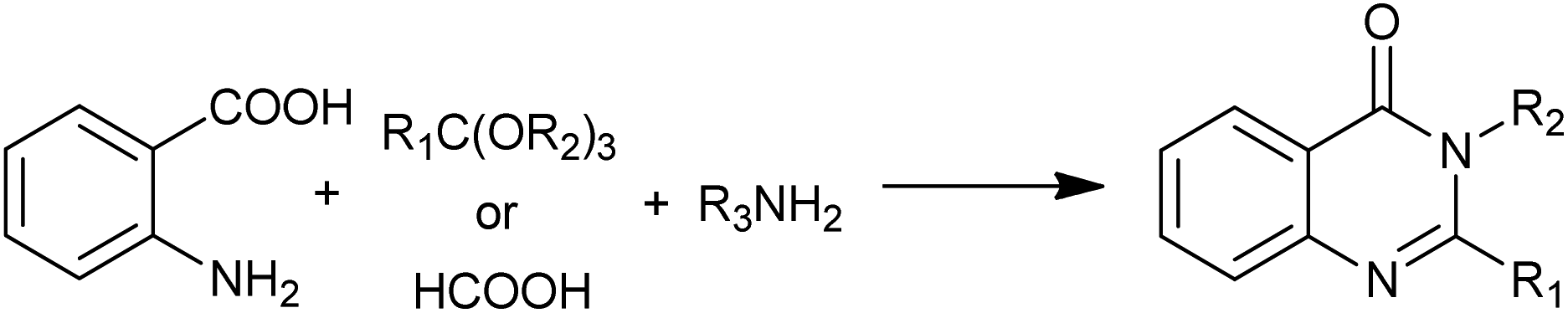DOI:
10.1039/C4RA00351A
(Review Article)
RSC Adv., 2014,
4, 12065-12077
Recent advances in 4(3H)-quinazolinone syntheses
Received
13th January 2014
, Accepted 18th February 2014
First published on 19th February 2014
Abstract
The 4(3H)-quinazolinone is a frequently encountered heterocycle with broad applications including antimalarial, antitumor, anticonvulsant, fungicidal, antimicrobial, and anti-inflammatory. The current review article will briefly outline the new routes and strategies for the synthesis of valuable 4(3H)-quinazolinones.
 Lin He | Lin He received her B.Sc. degree from Lanzhou University in 2005 and Ph.D. degree at Fudan University in 2013. She is currently a postdoctoral fellow in the Leibniz Institute for catalysis. Now, her major research focuses on carbonylative transformations. |
 Haoquan Li | Haoquan Li was born in 1988 in Zhongshan (P. R. China) and he studied chemistry in Wuhan at the Huazhong University of Science and Technology from 2007 to 2011 (supervised by Prof. Yanlong Gu). In 2013, he obtained a master's degree from Université de Rennes 1 under the supervision of Dr Jean-Baptiste Sortais and Prof. Christophe Darcel on iron catalysis. In July, 2013, he started his Ph.D. study at Leibniz-Institut für Katalyse (Rostock) supervised by Prof. Dr Matthias Beller and Prof. Dr Xiao-Feng Wu on the topic of palladium-catalyzed carbonylative coupling reactions. |
 Jianbin Chen | Jianbin Chen received his bachelor's degree in science in 2010 and earned a master's degree in 2013 (China). Since October 2013, he joined the group of Xiao-Feng Wu at Leibniz-Institute for Catalysis (Germany) for his Ph.D. study. |
 Xiao-Feng Wu | Xiao-Feng Wu studied chemistry at Zhejiang Sci-Tech University (China), where he got his bachelor's degree in science (2007). In the same year, he went to Rennes 1 University (France) and earned his master's degree in 2009, He completed his Ph.D. thesis in January 2012 under the supervision of Professor Matthias Beller at Leibniz-Institute for Catalysis (Germany). Then he started his independent research at ZSTU and LIKAT, and was promoted to Professor in 2013 in ZSTU. His research interests include carbonylation reactions, heterocycle synthesis, and the catalytic application of cheap metals. He also was a fellow of the Max-Buchner-Forschungsstiftung. |
1. Introduction
4(3H)-Quinazolinone and its derivatives constitute an important class of fused heterocycles that are found in more than 100 naturally occurring alkaloids.1 From a synthetic point of view, the first 4(3H)-quinazolinone compound 1 was obtained as early as 1869 from anthranilic acid and cyanogen.2 Intense search for biologically active substrate in this series was stimulated in the early 1950s with the elucidation of an alkaloid, febrifugine 2, which is an ingredient of a traditional Chinese herbal remedy, effective against malaria.3 Meantime (in 1951), methaqualone 3 was synthesized and it is the most well-known 4(3H)-quinazolinone based drug.4 It has sedative and muscle relaxant effects, although it causes photosensitization as a side effect (the marketing of methaqualone was terminated in 1984). At the moment, several marketed drugs5 which function as hypnotic/sedatives contain a 4-quiazolinone core. The use of 4(3H)-quinazolinone derivatives has also been proposed in the treatment of cancer. As the actual example, raltitrexed 4 (brand name: Tomudex), which launched in 1996 by Astra-Zeneca, is an antimetabolite drug used in cancer chemotherapy (Fig. 1).
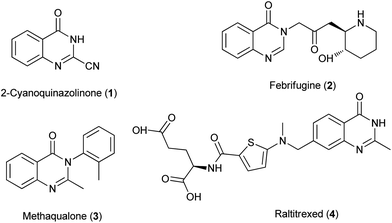 |
| | Fig. 1 Synthetic and natural quinazolinones. | |
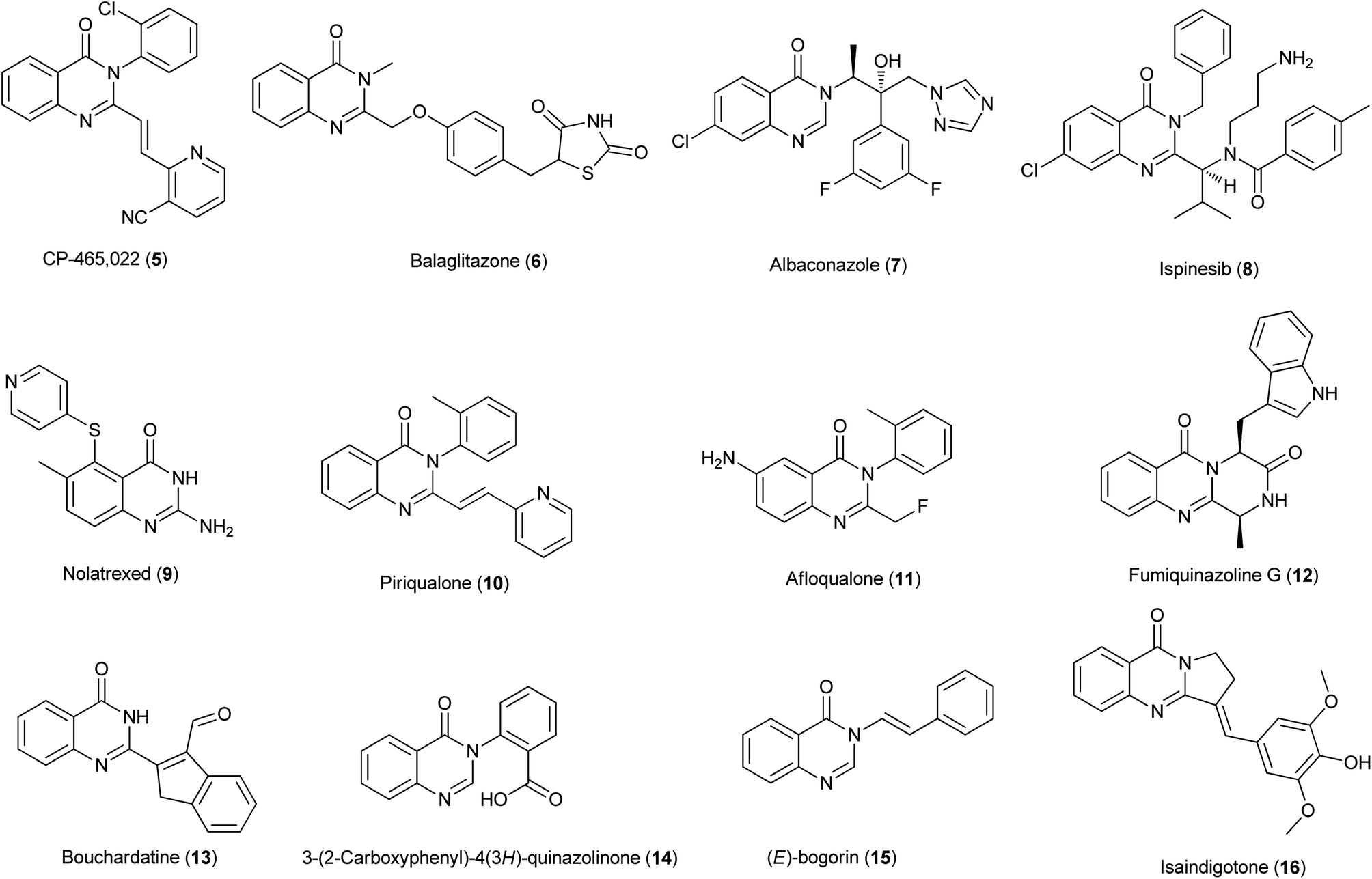
The 4(3H)-quinazolinone systems are now known to have a wide range of useful biological properties, e.g., anticancer, antiviral, anti-inflammatory, anti-microbial cholinesterase inhibitor, antifolate, antitumor, protein kinase inhibitor and many others.6 A few illustrative examples of quinazolinones that show potentially pharmacological activities7,8 are listed in Fig. 2. Regarding the importance of 4(3H)-quinazolinones, many useful synthetic procedures have been developed for their preparation. The comprehensive review by Guiry has covered the literature about methods for the construction of the 4(3H)-quinazolinones up to the end of 2004.9 Eguchi summed the chemistry of bioactive quinazolinone-based natural alkaloids in 2006.10 After that, relatively few reviews have appeared on 4(3H)-quinazolinone synthesis. We are focusing on carbonylative synthesis of heterocycles and the carbons in quinazolinones are potentially can be introduced by carbonylations. Therefore, we try to give an overview of the synthetic routes and strategies recently reported for the generation of highly functionalized 4(3H)-quinazolinones.
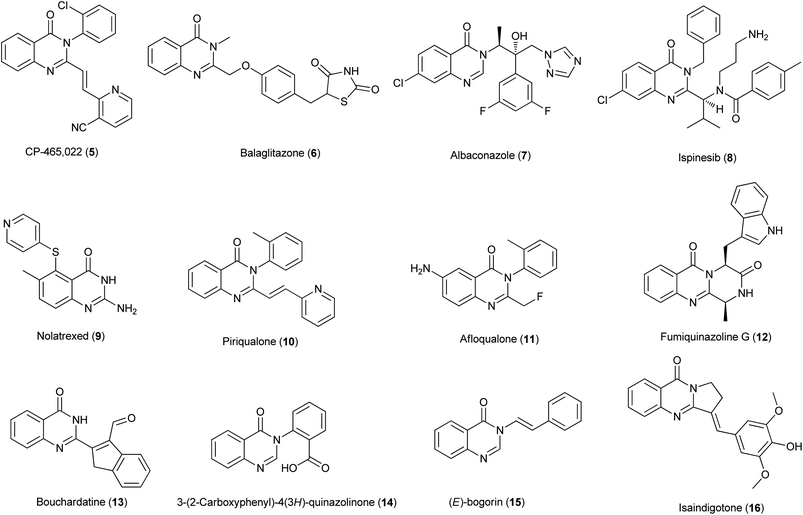 |
| | Fig. 2 Selected examples of quinazolinones with pharmacological activities. | |
2. Synthesis of 4(3H)-quinazolinone and derivatives
2.1 Niementowski quinazolinone synthesis
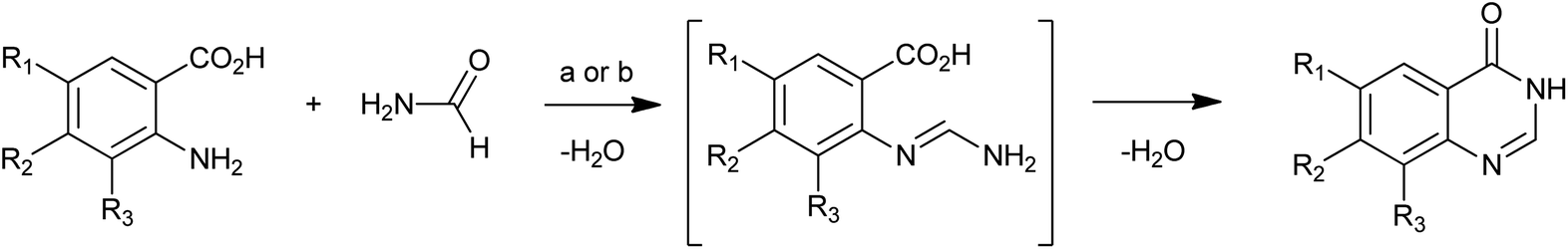
The most common method for 4(3H)-quinazolinone synthesis is based on the Niementowski reaction by the fusion (130–150 °C) of anthranilic acid analogues with amides, proceeding via an o-amidobenzamide intermediate (Scheme 1). The reaction yield in such conditions is variable and sometimes low yields were observed accompanied by complicated mixtures of carbonaceous compounds and impurities which were difficult to eliminate, even by column chromatography or recrystallization. To overcome these problems, Besson et al.11 have re-investigated the Niementowski synthesis of the 4(3H)-quinazolinone using microwave irradiation and have improved the yields and reduced the reaction time. By using microwave techniques, new efficient routes to novel fused quinazolinones (Scheme 2) have been developed.12,13
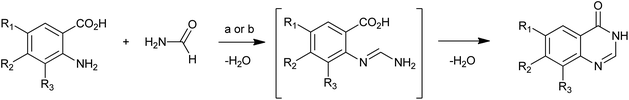 |
| | Scheme 1 The Niementowski reaction: (a) 130–150 °C, 6 h; (b) Besson's microwave conditions: MW (60 W), 20 min. | |
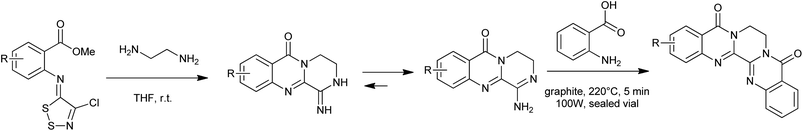 |
| | Scheme 2 Microwave-assisted Niementowski reaction for the synthesis of novel pentacyclic heterocycles. | |
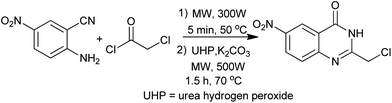
Desai et al. extended the microwave-assisted Niementowski reaction to synthesize 3-substituted/2,3-disubstituted-4(3H) quinazolinones instead of the 2-substituted derivatives. The methodology is environmentally benign and completely eliminates the need of solvent for the reaction.14 Vanelle et al. have investigated the preparation of new anticancer agent precursors in a quinazoline series.15 The Niementowski reaction could be easily and rapidly performed, affording the intermediate 2-chloromethyl-6-nitroquinazolin-4(3H)-one in good yield (Scheme 3). This compound served as a substrate for the synthesis of several new quinazolines, bearing various substituents in position 2.
 |
| | Scheme 3 The preparation of 2-substituted quinazolines. | |
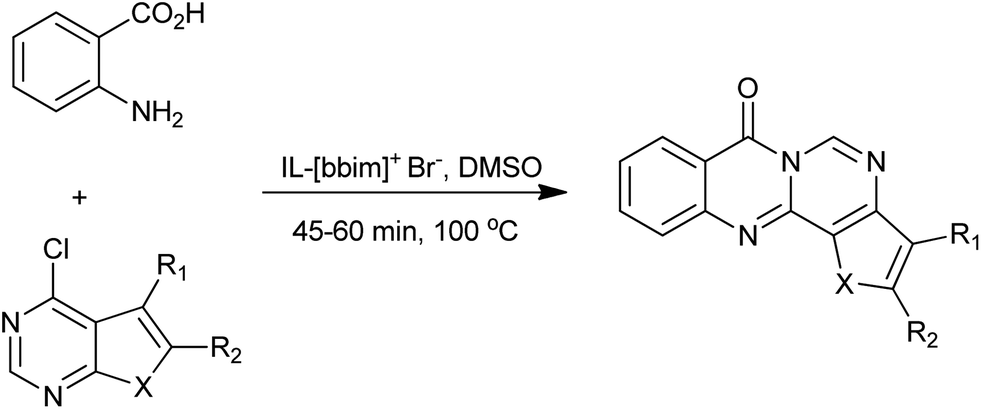
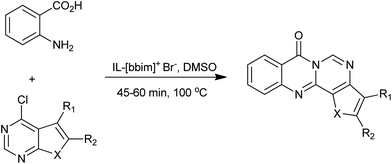
In 2011, Srinivasan et al. reported a new, rapid and a versatile approach using DMSO and ionic liquid as a chemical catalysts for the Niementowski synthesis of fused 4(3H)-quinazolinone (Scheme 4).16 High to excellent isolated yields (83–92%) were obtained with easy workup procedure. The enhanced reactivity was attributed to the inherent Brönsted acidity of ionic liquid and high polarity of both IL and DMSO.
 |
| | Scheme 4 Niementowski synthesis of modified quinazolinones. | |
2.2 Aza-Wittig methodology in 4(3H)-quinazolinone synthesis
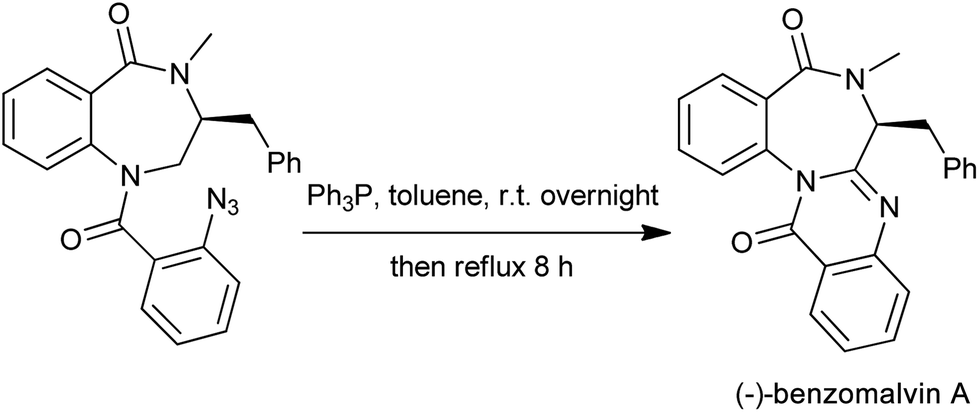
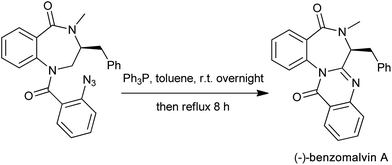
The aza-Wittig reactions of iminophosphoranes (Eguchi protocol) have become a powerful tool towards the construction of nitrogen heterocyclic compounds. Following the tandem aza-Wittig/cyclization strategy, various 2-substituted quinazolinones are synthesized starting from functional iminophosphorane bearing an amido group. A successful simple application of the aza-Wittig methodology is the convergent total synthesis of (−)-benzomalvin A,17 a microbial broth for neurokinin receptor antagonists. As the final drive to (−)-benzomalvin A, the iminophosphorane reacted with the imide carbonyl function to afford the desired product in 80% overall yield (Scheme 5).
 |
| | Scheme 5 The final step for the (−)-benzomalvin A synthesis. | |
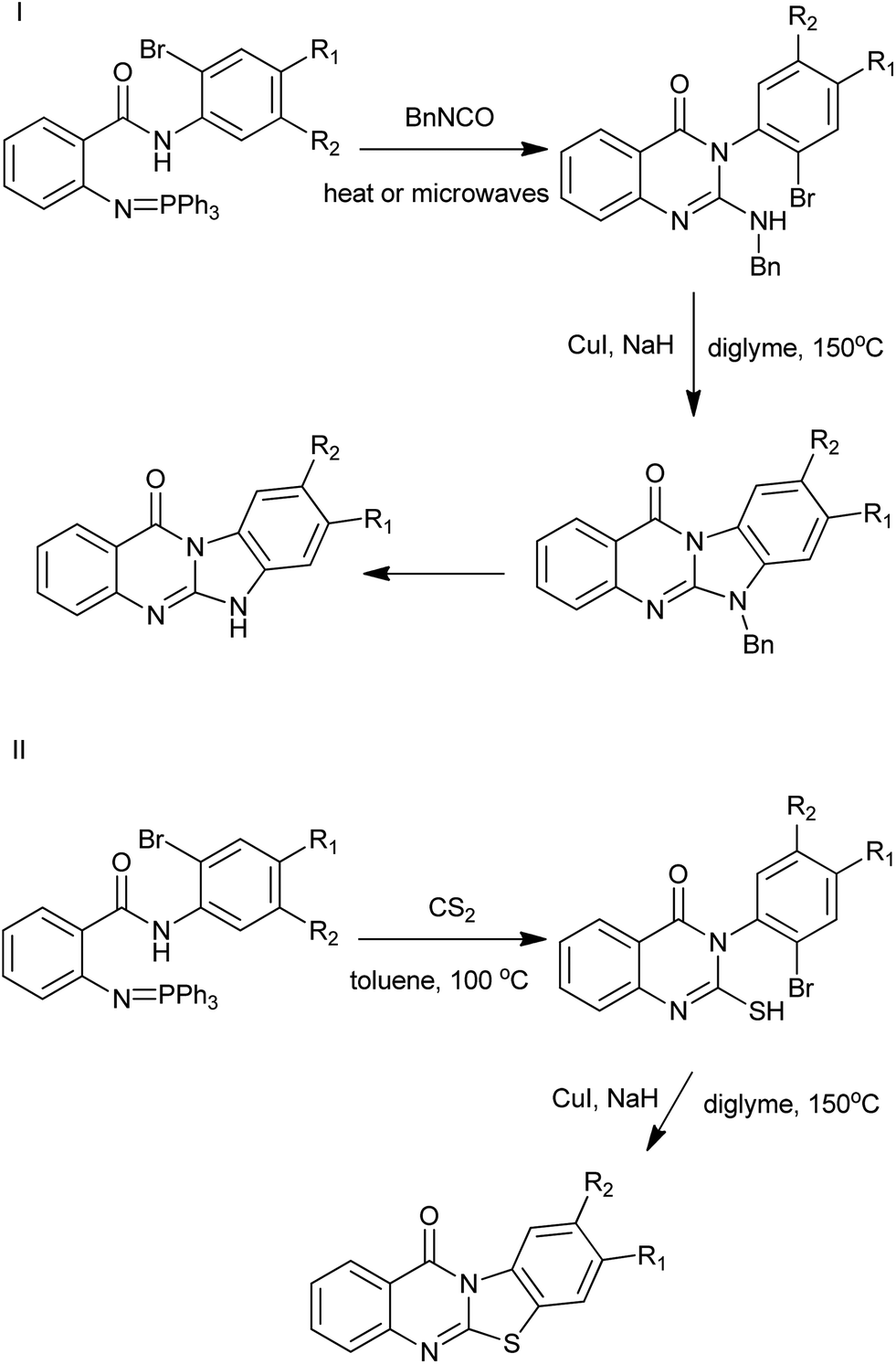
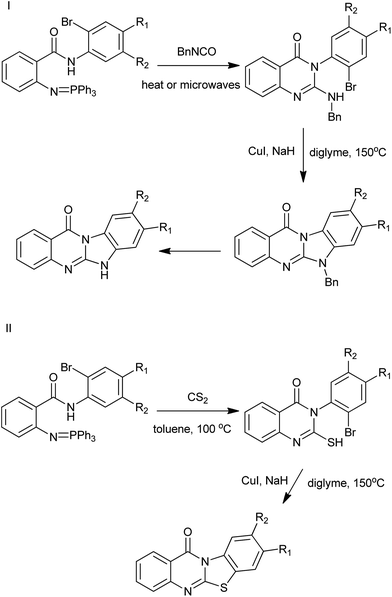
By combinations of aza-Wittig methodologies and CuI-catalysed heteroarylation processes, Molina et al. have developed an efficient route for the preparation of the not readily available linear or angular tetracyclic ring systems incorporating quinazolinone moieties (Scheme 6).18 The approach is based on the use of (arylimino)phosphoranes bearing N-(o-substituted-aryl)carboxamide substituents at their ortho positions. The presence of nitrogen functionality in the N-aryl substituent of promotes heterocyclization after an aza-Wittig reaction/reductive process. Iminophosphoranes also reacted in aza-Wittig-type fashion with carbon disulfide to provide functionalized aryl isothiocyanates.
 |
| | Scheme 6 Preparation of fused tetracyclic quinazolinones. | |
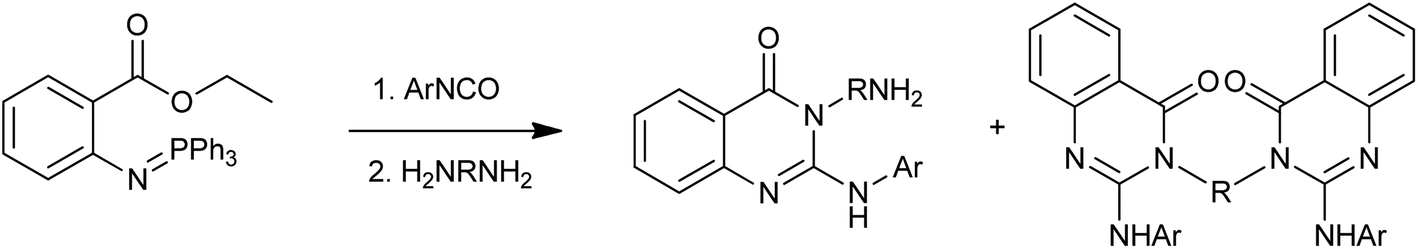
A fundamental approach has been reported by the Wu group for the synthesis of novel 3-aminoalkyl-2-arylaminoquinazolin-4(3H)-one and 3,3′-disubstituted bis-2-arylaminoquinazolin-4(3H)-ones via a tandem aza-Wittig reaction of 1-aryl-3-(2-ethoxycarbonylphenyl)carbodiimides with primary diamines (Scheme 7).19 The benefits of the method include mild reaction conditions, high selectivity, good yields, easily accessible starting materials and straightforward product isolation.
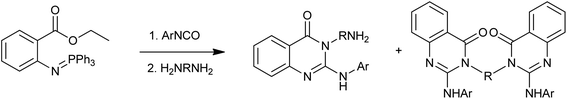 |
| | Scheme 7 The synthesis of 3-aminoalkyl-2-arylaminoquinazolin-4(3H)-one and 3,3′-disubstituted bis-2-arylaminoquinazolin-4(3H)-one. | |
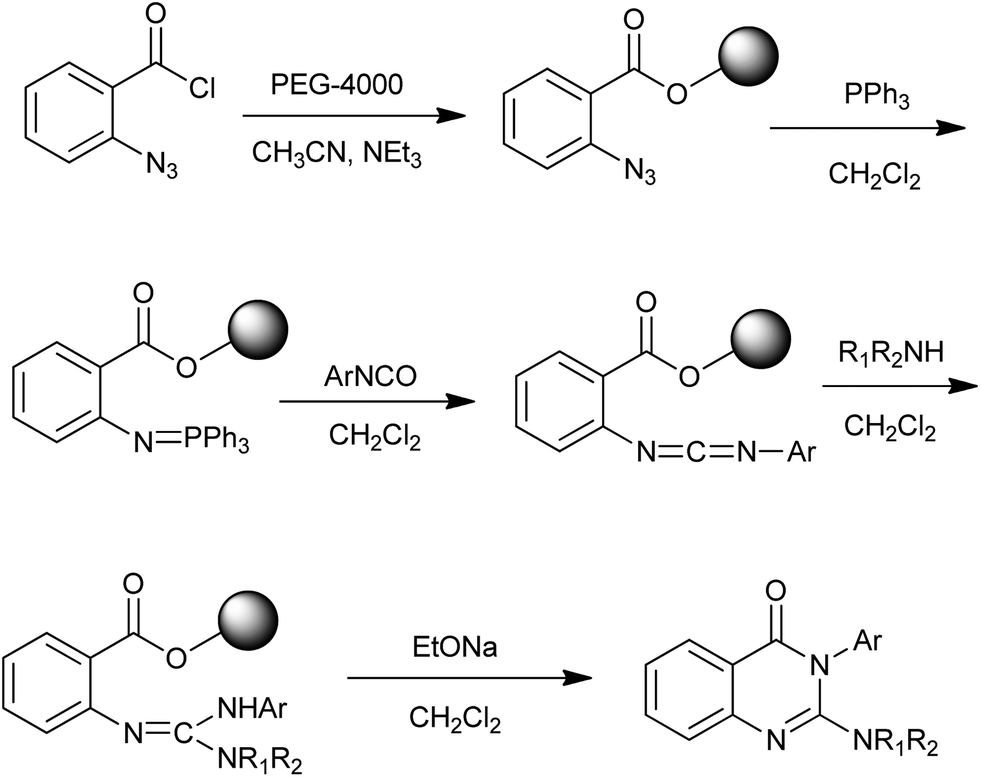
The use of polymer-supported reagents in organic synthesis effectively reduces workup and purification to a simple filtration. To date, a number of solid-phase approaches to quinazolinones have been developed and the related literature has been reviewed by Vögtle and Marzinzik in 2006.20 Recently, Ding et al. reported an efficient synthesis of 4(3H)-quinazolinones in parallel fashion via poly(ethylene glycol) (PEG) supported aza-Wittig reaction (Scheme 8).21 Commercially available cheap difunctional PEG-4000 was chosen as a soluble polymer support. A variety of secondary amine and isocyanate could be used for this synthetic strategy and the products were obtained in good yields.
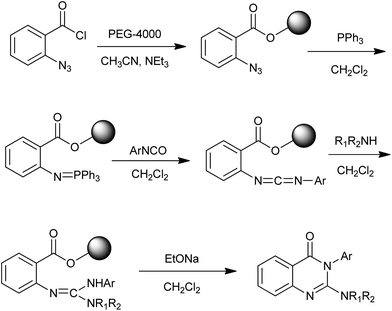 |
| | Scheme 8 Polymer-supported reagents for the synthesis of 4(3H)-quinazolinones. | |
2.3 Quinazolinone synthesis via benzoxazinone intermediate
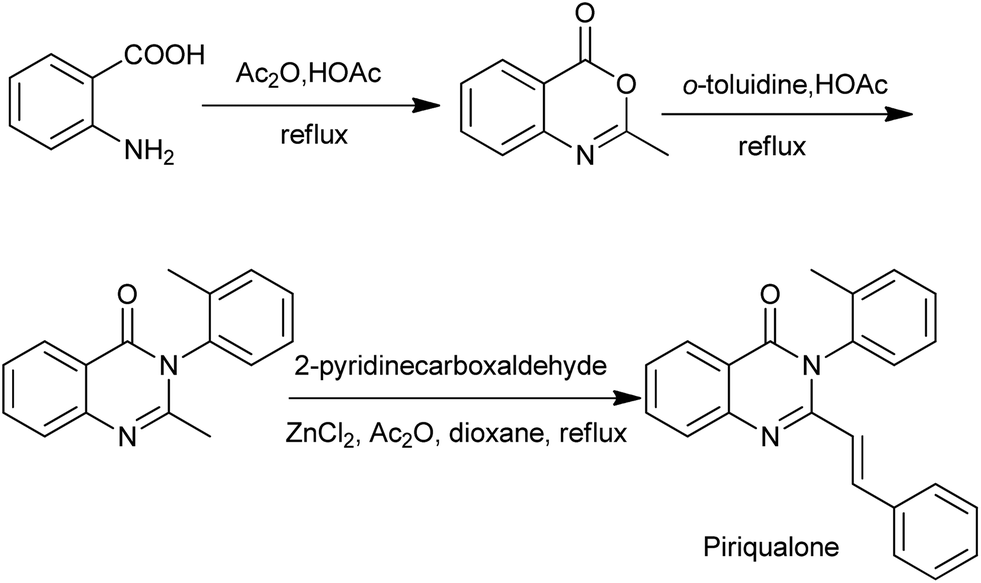
During the last decade, 4(3H)-quinazolinone syntheses via benzoxazinone intermediates have become very popular. Chenard et al. has described a succinct SAR program that led to the discovery of piriqualone CP-465022, a potent antagonist that interacts with the receptor through an allosteric site. The synthetic route to piriqualone (Scheme 9)22 combines three steps: anthranilic acid is converted to the benzoxazin-4-one with hot acetic anhydride in acetic acid. Subsequent reaction with a suitable aniline fragment in refluxing acetic acid installed the ortho toluidine ring. Finally, condensation of this 2-methyl-3-aryl-quinazolin-4-one with pyridine-2-carboxaldehydes provided the target compounds.
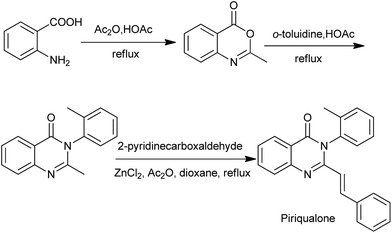 |
| | Scheme 9 The synthetic route to piriqualone. | |
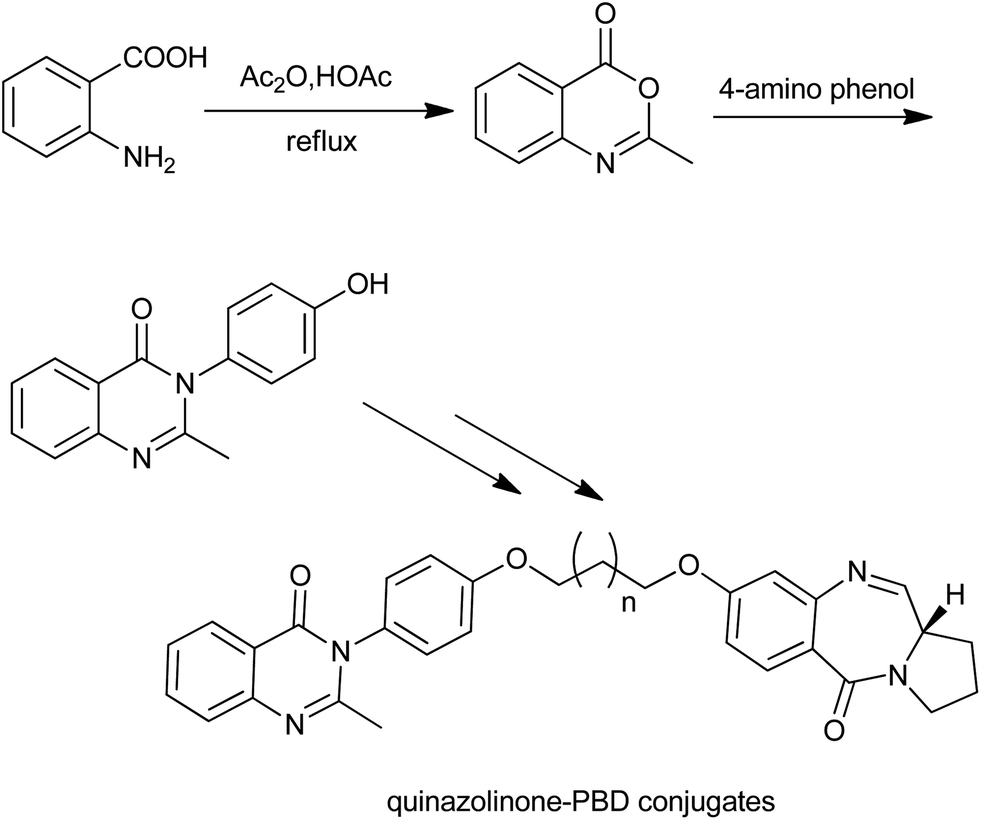
Similarly, Kamal et al. synthesized a series of novel quinazolinone linked pyrrolobenzodiazepine (PBD) conjugates (Scheme 10).23 The first synthetic step involved the condensation of 5-hydroxy anthranilic acid with acetic anhydride to afford benzoxazinones. By using this methodology, they also prepared a series of methaqualone analogs and 3-diarylethyne quinazolinones (a new class of senescence inducers).
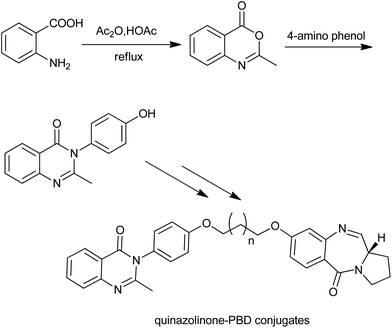 |
| | Scheme 10 The initial steps for the synthesis of quinazolinone linked pyrrolobenzodiazepine (PBD) conjugates. | |
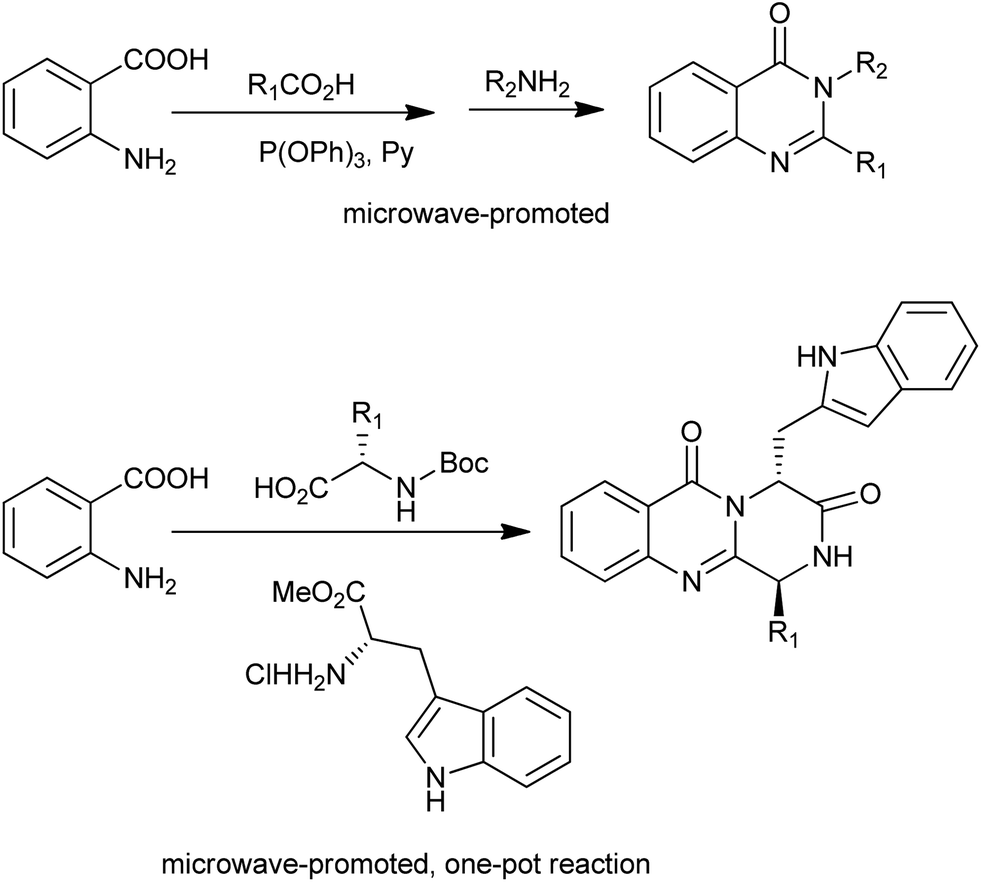
A microwave-promoted synthesis of 2,3-disubstituted 3H-quinazolin-4-ones with broad scope was developed by Liu and co-workers.24 The key step is the one-pot, two-step reaction sequence combining anthranilic acids, carboxylic acids, and amines providing efficient access to the desired heterocycles (Scheme 11). They further designed a microwave-promoted three-component one-pot reaction for the synthesis for the pyrazino[2,1-b]quinazoline-3,6-dione scaffold (Scheme 11),25 which is common to several families of alkaloids with significant biological activities.
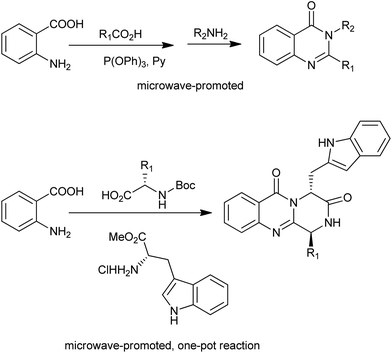 |
| | Scheme 11 Microwave-promoted synthesis of quinazolinones via benzoxazinone intermediate. | |
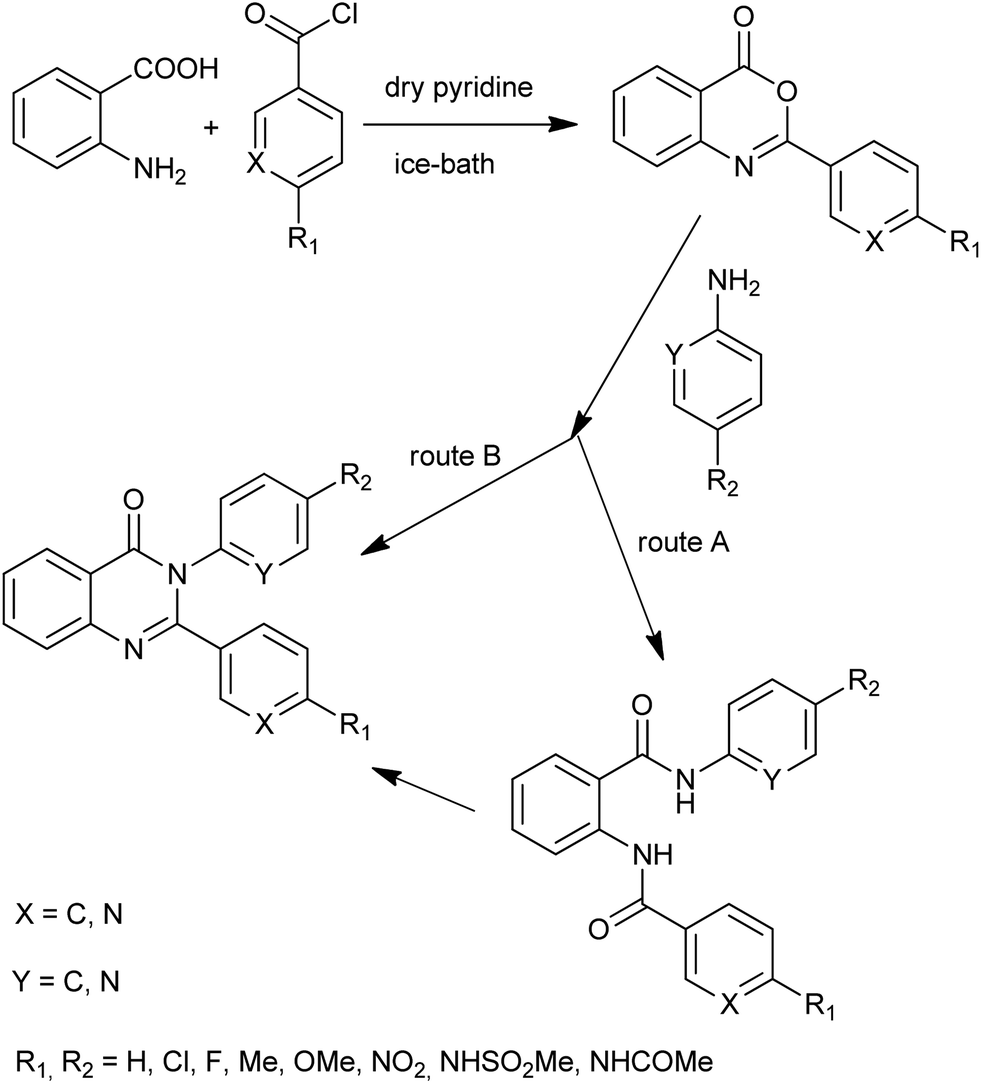
A new series of 3-arylquinazolines was prepared by Güngör et al.26 Although compounds of general formula were prepared by different methods depending on the nature and the positions of the substituents, most of them were obtained via benzoxazinone intermediate. Giridhar et al. synthesized various 2,3-diaryl-4(3H)-quinazolinones and evaluated for their anti-inflammatory activity (Scheme 12).27 Interestingly, the reaction of 2-phenyl-3,1-benzoxazin-4(3H)-one and 2-aminopyridine yielded diamide under milder conditions (at lower temperatures, 100 °C), while at higher temperatures (200 °C) the cyclized product 4(3H)-quinazolinone was isolated (route B). This observation is restricted to compounds having the pyridyl ring either in the 3,1-benzoxazin-4(3H)-one component or in amine component. For their cyclization into the final quinazolinones, a catalytic amount of anhydrous zinc chloride (route A) was used.
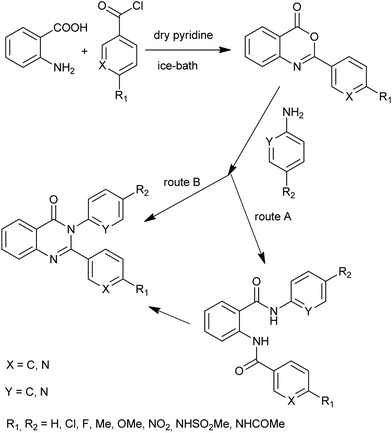 |
| | Scheme 12 2,3-Diaryl-4(3H)-quinazolinones synthesis. | |
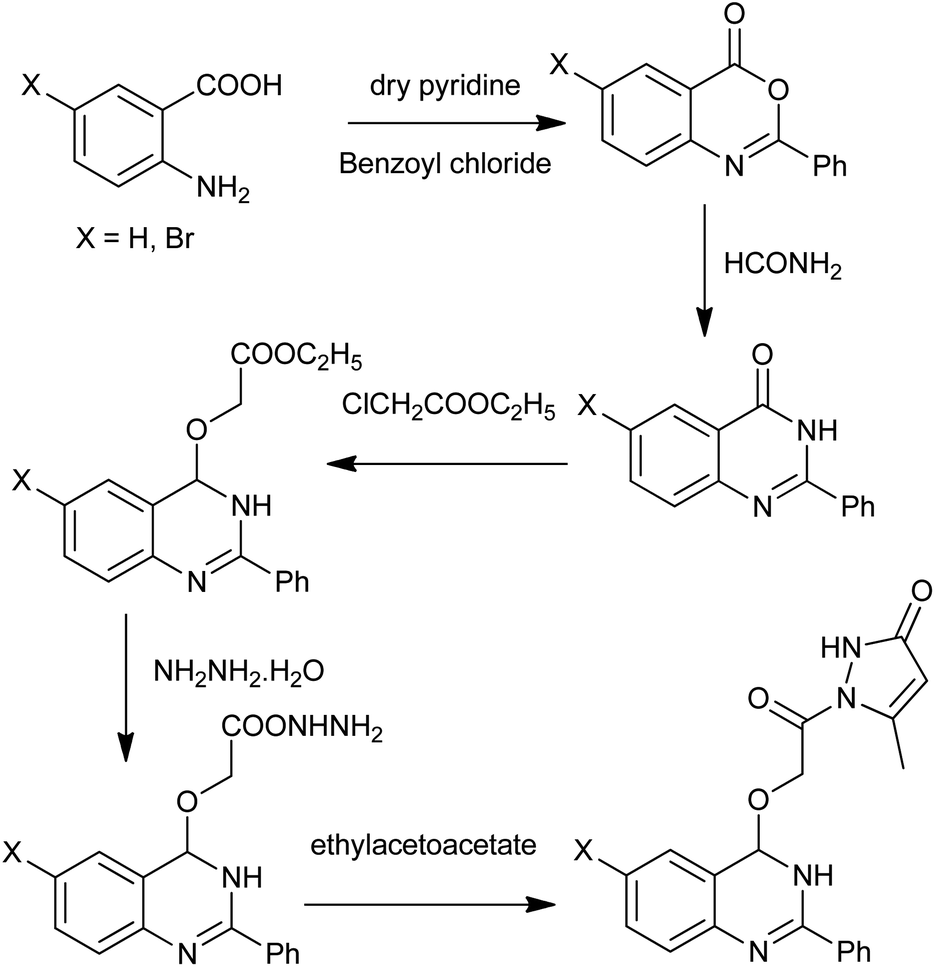
For the design of newer antibacterials, Boyapati et al. described the synthesis of novel quinazolinones with substitution of OCH2CONH2 (oxymethylcarbamide) group at the 4th position (Scheme 13),28 which were known to enhance the biological activities. For the preparation of desired products, anthranilic acid underwent cyclization with benzoyl chloride in pyridine at 0–5 °C to give 2-phenyl-(4H)-3,1-benzoxazin-4-one. Then, the obtained intermediate, on the reaction with formamide, results in the more stable 2-phenyl-4(3H) quinazolinone.
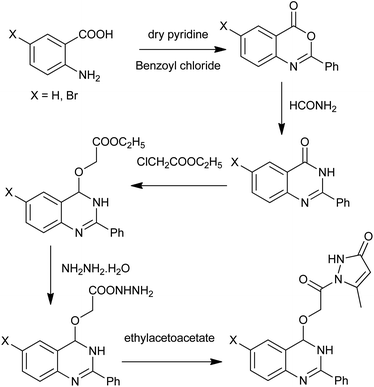 |
| | Scheme 13 Synthesis of 4-substituted quinazolines. | |
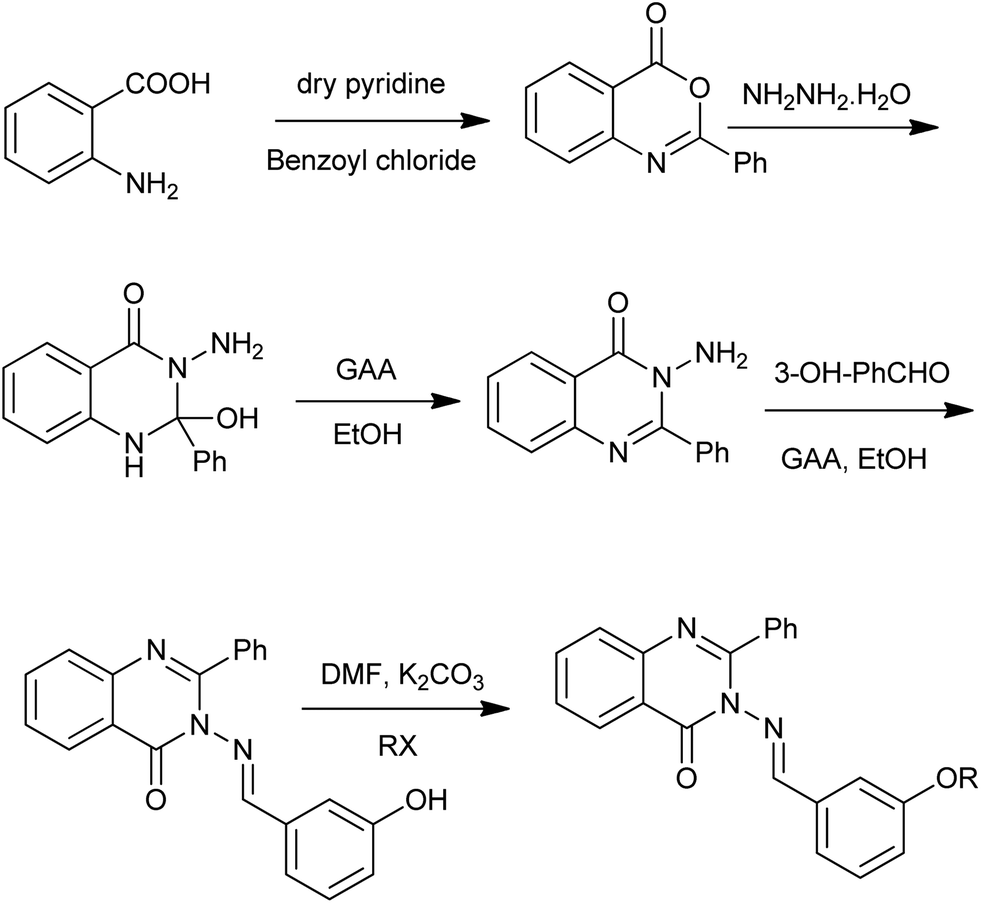
In 2013, Gupta et al. reported the synthesis and biological evaluation of some new quinazolin-4(3H)-ones derivatives (Scheme 14) as anticonvulsants.29 The first step is the condensation of anthranilic acid and benzoyl chloride in the presence of pyridine. The treatment of the obtained 2-phenyl-benzo[d][1,3]oxazin-4-one with hydrazine hydrate yielded 3-amino-2-phenyl-1H-quinazolin-4-one. Using the similar method, Alagarsamy and Saravanan obtained eighteen novel quinazolin-4(3H)-one derived pyrazole analogs.30
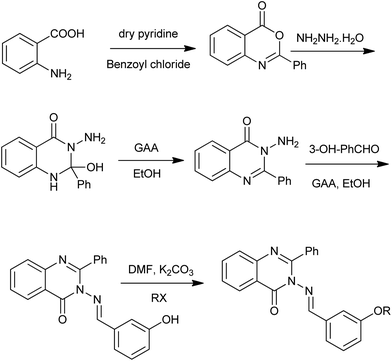 |
| | Scheme 14 Synthesis of quinazolinone derivatives. | |
2.4 Cyclocondensation of anthranilic acid, ortho esters (or formic acid), and amines
As a type of multi-component reactions (MCRs), one-pot condensation of anthranilic acid, ortho esters (or formic acid) and amines is one of the most straightforward procedures for the preparation of 4(3H)-quinazolinones (Scheme 15). Different acid catalysts are known to affect this condensation. Khosropour et al. have demonstrated Bi(TFA)3 immobilized on [nbp]FeCl4 could catalyzed the condensation with high to excellent yields of the 4(3H)-quinazolinones.31 The notable features of this procedure are mild reaction conditions (room temperature), clear reaction profiles, improved yields for both anilines and primary amines, enhanced rates and simplicity in operation. Moreover, the reusability, stability and non-toxicity of the catalyst and ionic liquid are other noteworthy advantages of this method.
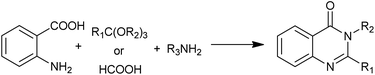 |
| | Scheme 15 One-pot condensation of anthranilic acid, ortho esters (or formic acid) and amines. | |
Hamdi et al. have found that the use of Keggin-type heteropolyacids (H3PW12O40·13H2O) coupled with microwave irradiation allows a solvent-free, rapid condensation reaction.32 Compared to the conventional method, the reaction time was ten-fold shorter (13 min for microwave irradiation versus 120 min for conventional heating). By using Keggin-type heteropolyacids as efficient, reusable, and eco-friendly heterogeneous inorganic catalysts, Heravi et al. developed a simple multi-component synthesis of 4-arylaminoquinazolines from the reaction of 2-aminobenzamide, ortho esters, and substituted anilines.33 As a related work, they also reported another method for the preparation 4(3H)-quinazolinones from reaction of 2-amino-benzamide, acylchlorides in the presence of catalytic amounts of (H14[NaP5W30O110])/SiO2 under ultra-sonic irradiation.34
Recently, Wang et al. found that SrCl2·6H2O could be used as an efficient and recyclable catalyst in one-pot condensation of anthranilic acid, ortho esters and amines leading to the formation of 4(3H)-quinazolinone derivatives in good yields at room temperature under solvent-free conditions.35 The method offers several advantages including simple work-up, mild conditions, commercially available catalyst, and the relatively clean procedure.
A new approach to the facile synthesis of 2-substituted-quinazolin-4(3H)-ones and its derivatives using the condensation reaction of substituted 2-aminobenzamide and ortho esters is reported by Huang in 2011.36 Interestingly, the reaction proceeds without organic solvent and in the absence of basic or acidic catalyst.
To elucidate the mechanism of action and investigate the drug metabolism of 4(3H)-quinazolinones, the preparation of suitable metabolically stable carbon-14 labels were required. Saemian et al. obtained the quinazolin-4(3H)-ones-[4-14C] from 2-aminobenzoic acid-[carboxy-14C], formic acid and amine.37 The reaction performed under microwave condition in the absence of solvent or any dehydrating agents.
2.5 Oxidative heterocyclization for 4(3H)-quinazolines synthesis
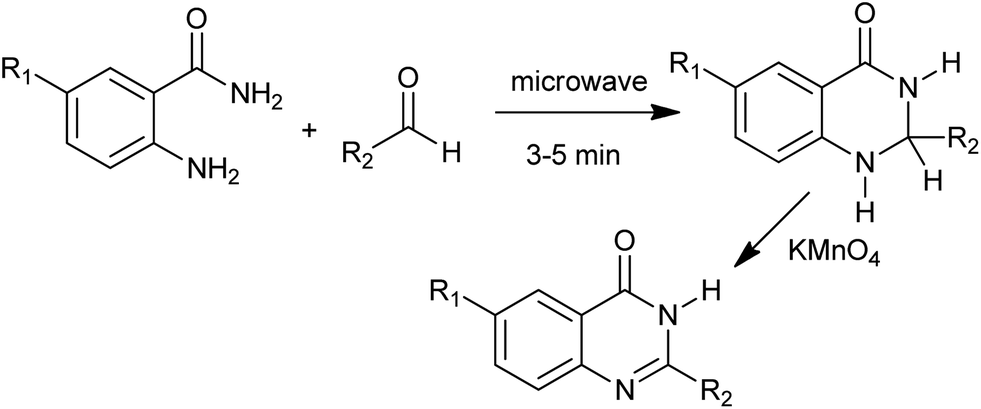
Bakavoli et al. have synthesized 4(3H)-quinazolines in one-pot by the oxidative heterocyclization of o-aminobenzamides with aldehydes in the presence of KMnO4 under microwave irradiation (Scheme 16).38 They also developed the I2/KI mediated oxidative cyclocondensation of o-amminobenzamide with various aldehydes.39 In ethanol–water or boiling water, the target 4(3H)-quinazolines formed in good to excellent yields.
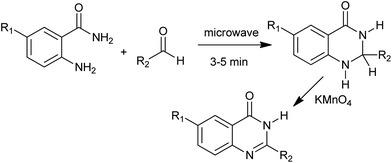 |
| | Scheme 16 Microwave activated oxidative heterocyclization. | |
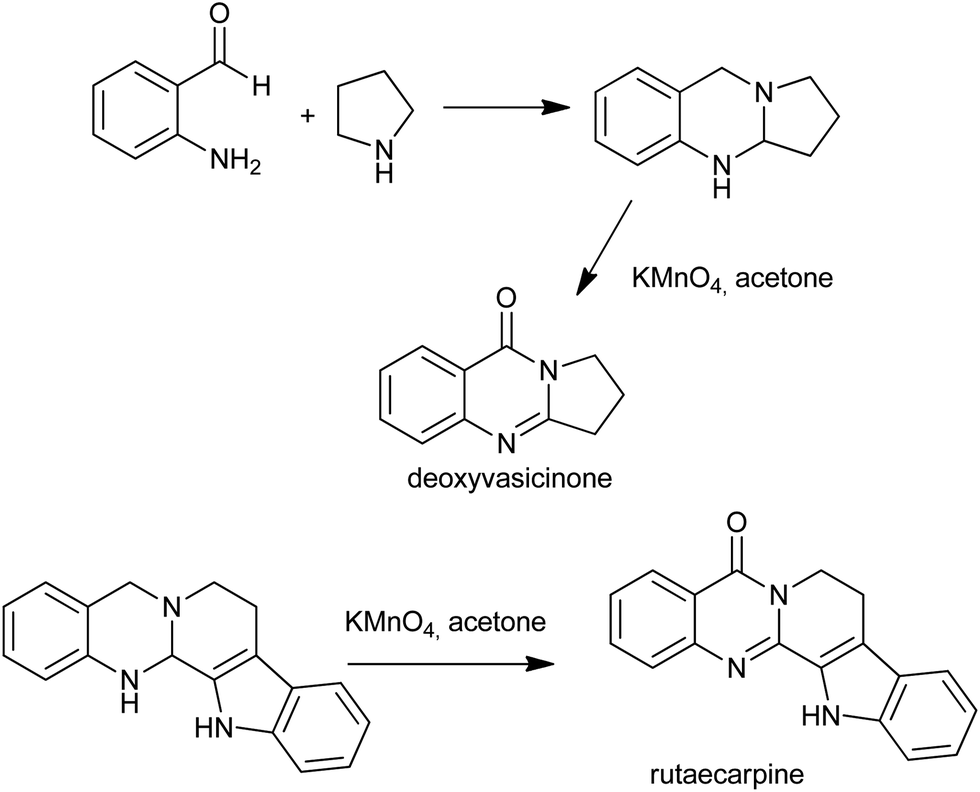
In 2008, Seidel et al. reported the syntheses of deoxyvasicinone and rutaecarpine by the potassium permanganate promoted oxidation of aminals, which in turn were obtained from the condensation of o-aminobenzaldehydes and simple secondary amines (Scheme 17).40 Later, the same group showed that quinazolinone alkaloids and their analogues can be synthesized from their corresponding aminals by using a copper(II) acetate/acetic acid/O2 and potassium iodide/tert-butylhydroperoxide systems.41 The use of acetic acid in addition to O2 and catalytic copper(II) salts was determined to prevent overoxidation of dihydroquinazolines, allowing access to these structures under mild conditions.
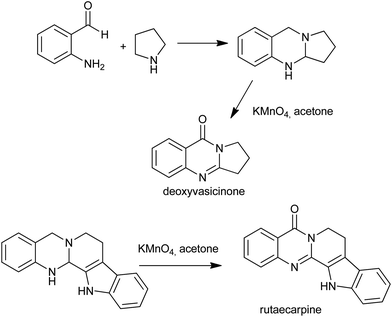 |
| | Scheme 17 The syntheses of deoxyvasicinone and rutaecarpine. | |
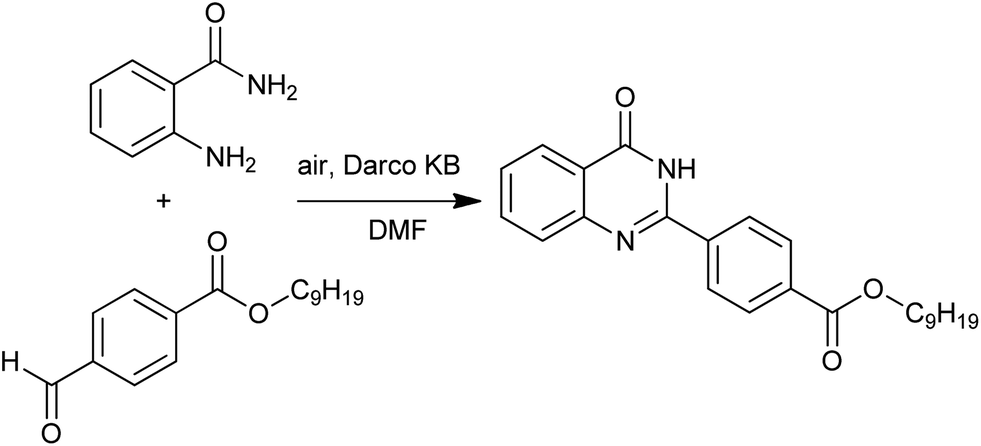
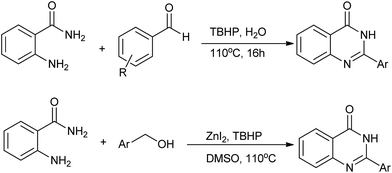
A solid-phase combinatorial synthesis of 2-arylquinazolines and 2-arylquinazolinones via oxidative coupling of aromatic aldehydes with 2-aminobenzylamines or 2-aminobenzamide was developed by Hioki and co-workers.42 Darco KB (a sort of activated carbon) was shown to be an effective catalyst for the air oxidation (Scheme 18).
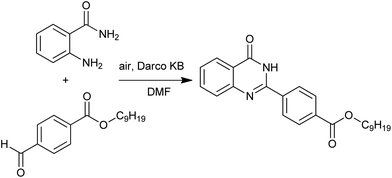 |
| | Scheme 18 Darco KB catalyzed oxidative heterocyclization. | |
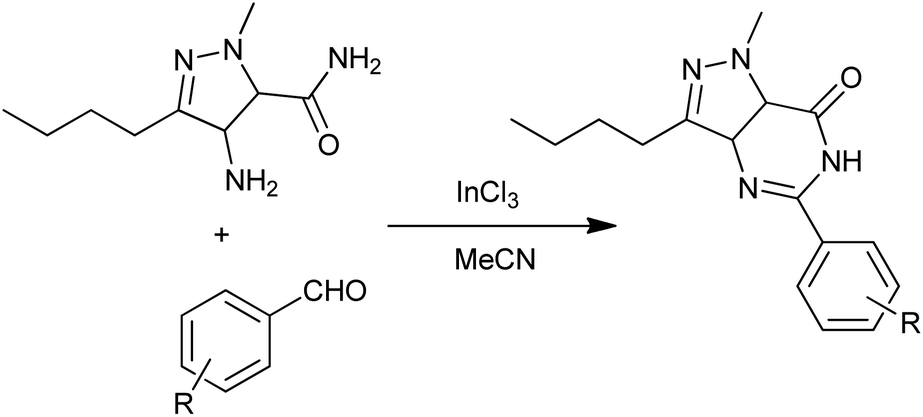
Mulakayala et al. reported a convenient and practical InCl3-catalyzed condensation of aromatic aldehydes with 2-aminobenzamides to quinazolinones (Scheme 19) in 2012.43 Compared to conventional Lewis acids, InCl3 has advantages of higher stability in water, operational simplicity, strong tolerance to oxygen and nitrogen containing substances and functional groups and it can often be employed in catalytic amounts.
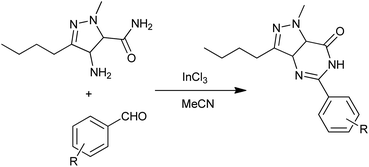 |
| | Scheme 19 InCl3-catalyzed synthesis of 5-aryl pyrazolo [4,3-d]pyrimidin-7(6H)-ones. | |
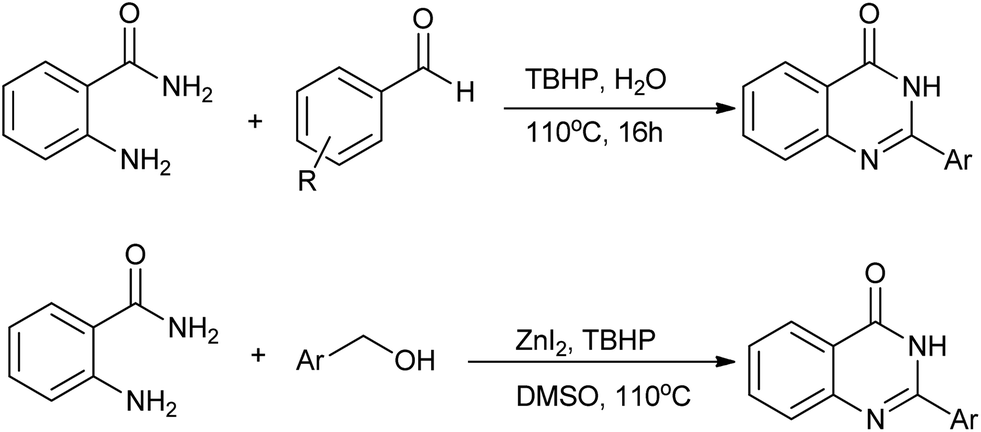
Very recently, our group reported an interesting procedure for quinazolinones synthesis by the reaction of 2-aminobenzamide with aldehydes under catalyst free conditions (Scheme 20).44 Halogen and electron-withdrawing functional groups in aromatic aldehydes are tolerable and gave the desired products in good to excellent yields. Several heterocyclic aldehydes were applied as substrates because of the interesting biological activities of heterocycles, the corresponding 2-heterocyclic substituted quinazolinones were synthesized straightforward in good yields. Even aliphatic aldehydes were reacted with 2-aminobenzamide as well and gave the corresponding alkyl-substituted products in good yields, which are difficult in previous methodology. Furthermore, we exploited the reactivity of stable alcohol for the synthesis quinazolinones via a zinc-catalyzed oxidation procedure. As expected, various desired products were prepared in good to excellent yields in one-pot one-step manner.
 |
| | Scheme 20 Oxidative synthesis of quinazolinones. | |
2.6 Synthesis of quinazolinones under transfer hydrogen conditions
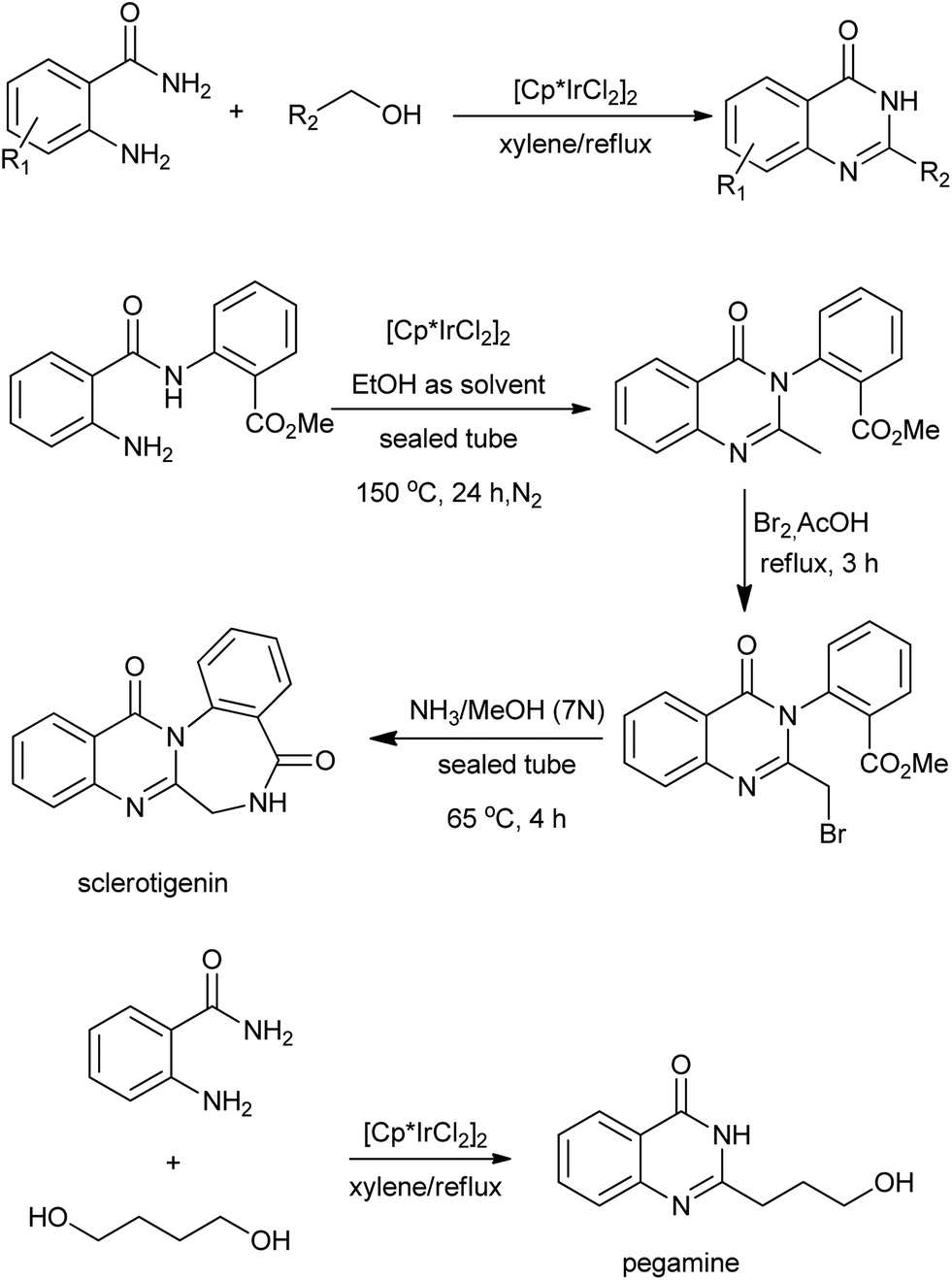
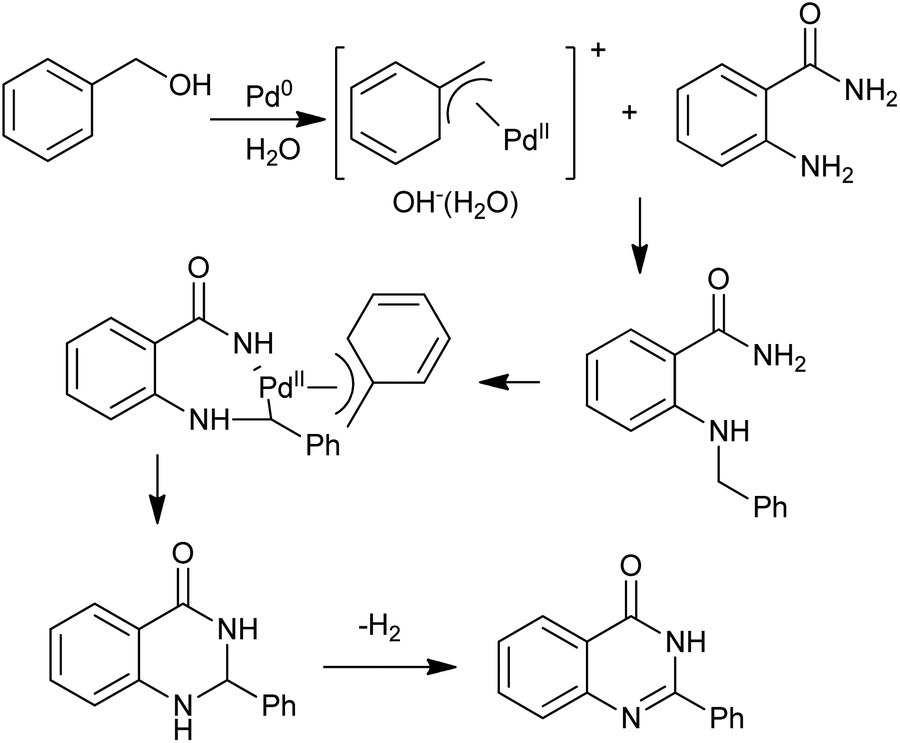
The N-alkylation of amines with alcohols via a hydrogen transfer strategy has become a useful tool for C–N bond formation. This domino reaction sequence is based on the dehydrogenation of the alcohol in situ to give the corresponding aldehyde or ketone. Subsequent imine formation followed by reduction with the hydrogen initially produced leads to the N-alkylated amine. Based on this methodology, Zhou et al. demonstrated a one-pot synthesis of quinazolinones between primary alcohols and o-aminobenzamides via Ir-catalyzed dehydrogenations under base-free conditions (Scheme 21).45 Notably, they extended this operationally convenient method towards the synthesis natural products including sclerotigenin and pegamine.46 A similar reaction for the synthesis of 4-phenylquinazolinones via a Pd-catalyzed domino reaction of o-aminobenzamides with benzyl alcohols were reported by Yokoyama and co-workers.47 A mechanism that quite different from Ir-catalyzed hydrogen transfers process was proposed. The oxidative additions of benzyl alcohols to Pd0 afford the (η3-benzyl)palladium complex in aqueous media, which plays an important role in the benzyl transfer and C–H activation (Scheme 22). Furthermore, subsequent reaction steps for C–H activation require an oxidant capable of converting palladium to a higher oxidation state, and benzyl alcohols work for regeneration of PdII species to toluene in the system.
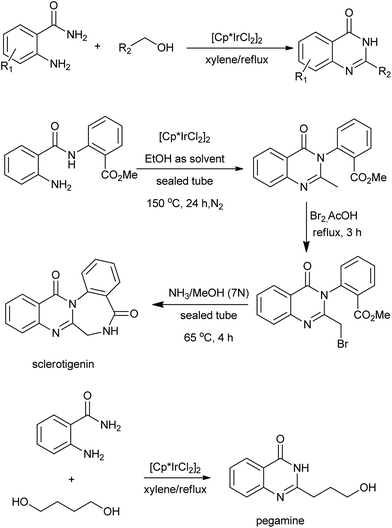 |
| | Scheme 21 Ir-catalyzed synthesis of quinazolinones between primary alcohols and o-aminobenzamides. | |
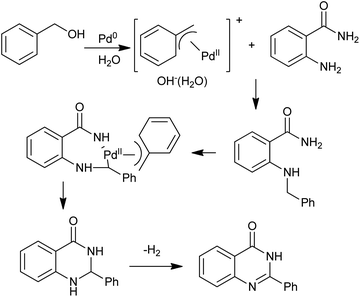 |
| | Scheme 22 Pd-catalyzed synthesis of quinazolinones starting with benzyl alcohol. | |
Usually, the amino group is prepared from the corresponding nitro group via a reduction process using a stoichiometric amount of metal/acid or hydrogen. As for the synthesis of quinazolinones, the direct use of commercially available and inexpensive nitroarenes and alcohols as starting materials is highly attractive. In this straightforward transformation, the alcohol conceivably might serve two possible functions: as hydrogen source for nitro reduction and as alkylating reagent based on the catalytic hydrogen transfer. In 2013, Deng et al. showed that a series of 2,3-diarylquinazolinones can be synthesized from nitrobenzamides and alcohols using nontoxic iron as catalyst (Scheme 23).48 Halogens as well as other active groups were well tolerated under the given reaction conditions.
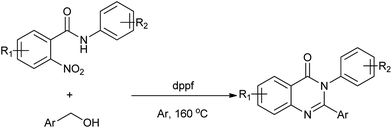 |
| | Scheme 23 Fe-catalyzed method for the synthesis of 2,3-diarylquinazolinones. | |
2.7 Formation of quinazolinones via radical cascades
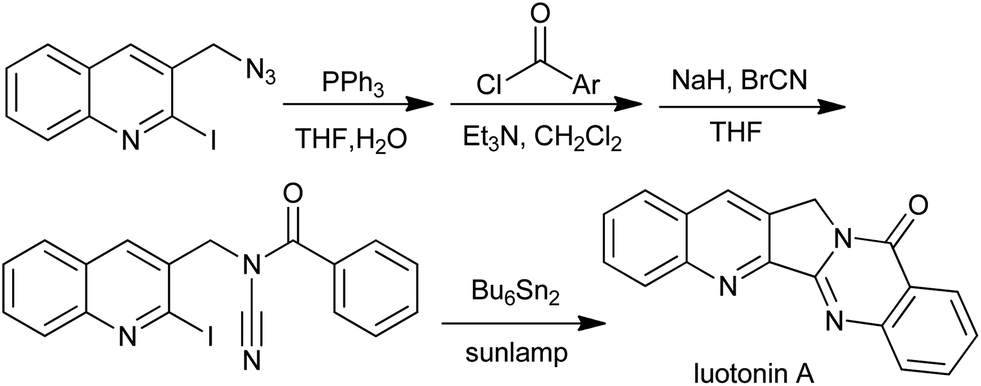
An elegant approach for the total synthesis of luotonin A based on radical cascade was reported by Malacria et al., involving N-acyl cyanamides (Scheme 24).49,50 A broad variety of pyrimidones fused with alkyl, aryl, or heteroaryl moieties can be prepared this way. A detailed mechanism study51 showed that the central feature of the reaction was the radical migration of hydrogen atoms or carbon substituents triggered by rearomatization of a cyclohexadienyl radical.
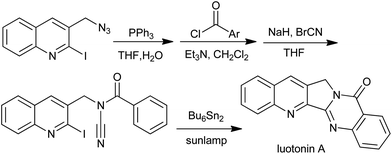 |
| | Scheme 24 Total synthesis of luotonin A. | |
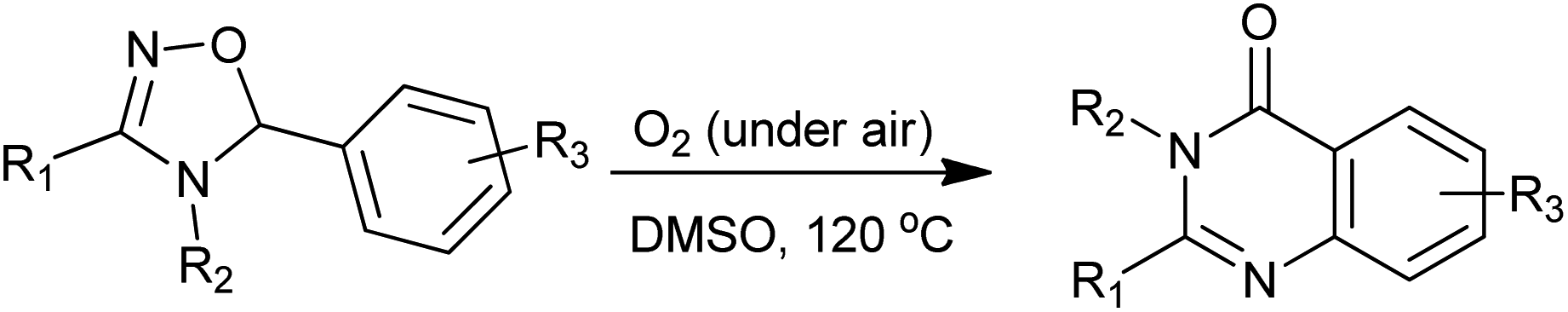
Chiba et al. developed a concise syntheses of biologically active quinazolinone derivatives via oxidative radical skeletal rearrangement of 5-aryl-4,5-dihydro-1,2,4-oxadiazoles induced by molecular oxygen (Scheme 25).52,53 The product selectivity was realized by the difference of the reaction temperature as well as the choice of solvents and inorganic bases. This atom- and step-economical approach allows the facile construction of quinazolinone cores under tin-free aerobic radical conditions.
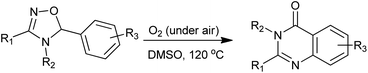 |
| | Scheme 25 Oxidative radical skeletal rearrangement of 5-aryl-4,5-dihydro-1,2,4-oxadiazoles into quinazolinones. | |
2.8 Transition metal-catalyzed N-arylation for quinazolinone synthesis
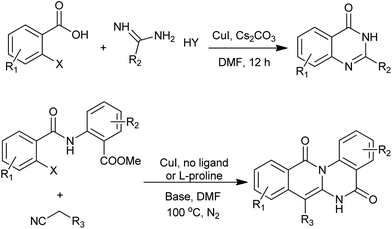
Transition metal-catalyzed catalyzed Ullmann N-arylations is considered to be an important strategy that finds wide applications in the synthesis of many substances. In this context, Fu et al. and co-workers have developed efficient copper-catalyzed N-arylations for the preparation of quinazolinone derivatives (Scheme 26).54 Notably, they demonstrated that the coupling reactions of 2-bromo- and iodobenzoic acid derivatives with amidines can be performed well at 25 °C without the addition of a ligand or an additive. Even nonactive substrates, such as 2-chlorobenzoic acid or guanidines, can be readily transformed into the corresponding quinazolinone in the presence of CuI at 80 °C. This method represents the first example of constructing N-heterocycles under ligand-free copper catalysis at room temperature. Following that, they described an iron-catalyzed cascade synthesis of 1,2,4-benzothiadiazine 1,1-dioxide and quinazolinone derivatives.55 The same group also reported a novel and useful domino method for construction of quinazolinones using readily available amino acids as the nitrogen-containing motifs.56 Most recently, they succeed in preparation of isoquinolino[2,3-a]quinazolinones via a copper-catalyzed method.57 The process comprises an Ullman-type C-arylation, intramolecular addition of NH with CN, and nucleophilic attack of amino to ester group.
 |
| | Scheme 26 Cu-catalyzed synthesis of quinazolinone derivatives. | |
An efficient iron-catalyzed synthesis of a series of quinazolinone derivatives in both H2O and DMF under microwave irradiation was developed by Liu and co-workers.58 This is the first report of iron-catalyzed C–N coupling forming N-heterocycles in aqueous media. Later, they also described an iron/copper co-catalyzed synthesis of 2-methylquinazolin-4(3H)-one under microwave irradiation.59
To meet the challenges of sustainable pathways for organic transformations, Wang et al. report an efficient magnetically recoverable and reusable Fe3O4 nanoparticle-supported copper(I) catalyst and its application in cascade reactions of amidines with substituted 2-halobenzoic acids and 2-bromocycloalk-1-enecarboxylic acids to synthesize quinazolinone and bicyclic pyrimidinone.60 Recovery of the catalyst by decantation of the reaction mixture in the presence of an external magnet is easy and efficient. The catalyst was recycled over 10 times in reactions without any obvious loss in its activity.
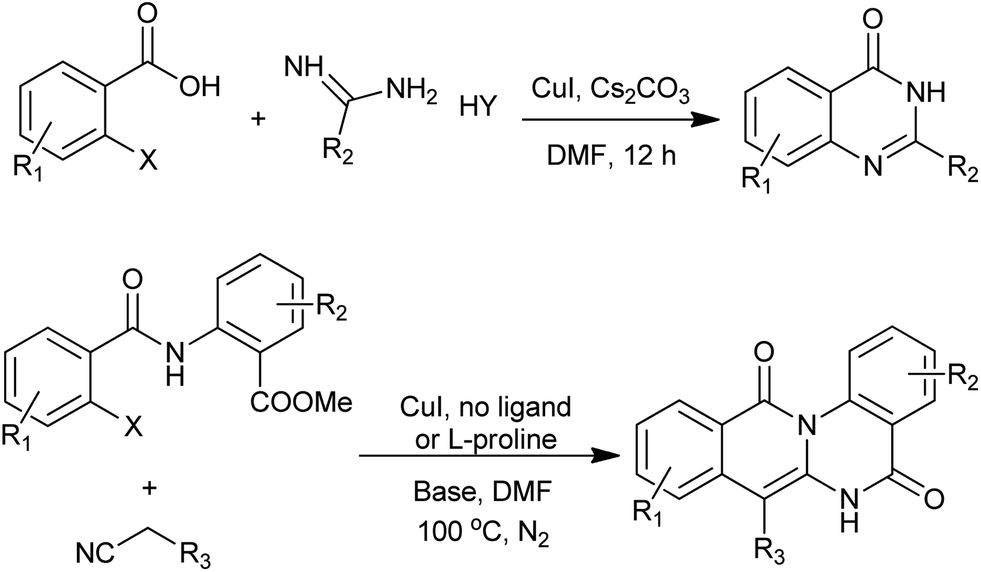
A facile and efficient approach for assembling substituted quinazolinones has been developed by Ma group (Scheme 27).61 Conveniently available amides could serve as suitable nucleophiles for coupling reactions with N-substituted o-bromobenzamides, affording 3-substituted and 2,3-disubstituted quinazolinones after spontaneous or HMDS/ZnCl2 mediated condensative cyclization. The usage of this protocol has been demonstrated by formal synthesis of dictyoquinazol A and preparation of methaqualone.
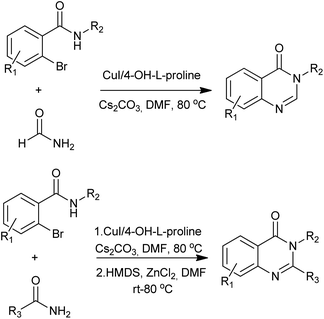 |
| | Scheme 27 Cu-catalyzed aryl amidation. | |
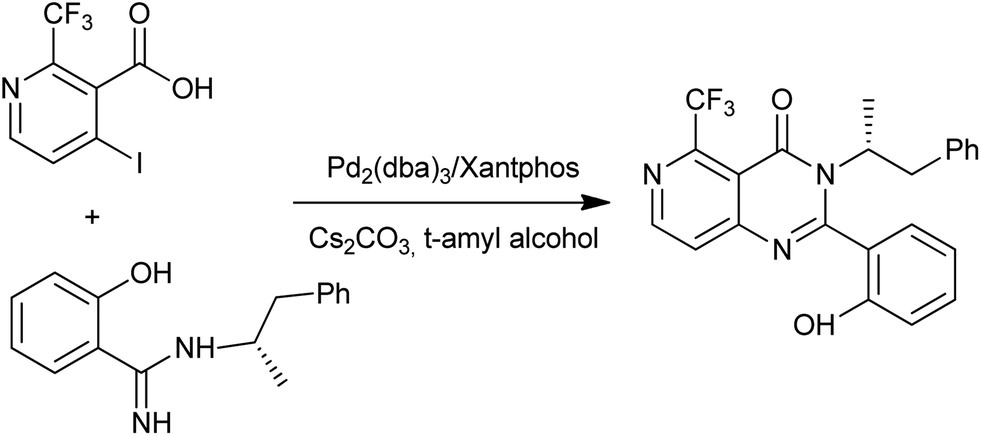
Li et al. were prompted to develop a scalable synthesis of pyrido[4,3-d]pyrimidin-4(3H)-one (Scheme 28), which is an orally active calcium-sensing receptor (CaR) antagonist targeted for the treatment of osteoporosis.62 The first attempt using CuI mediated conditions gave the desired product; however, the reaction proceeds to 50% conversion at best, despite all optimization efforts with or without ligands used in the reaction. They quickly switched over to Pd-catalyzed methods and found the Pd2(dba)3/Xantphos system gave complete conversion and afforded a clean reaction profile by HPLC analysis. The amidine N-arylation worked well with 2-bromo or iodo benzoate methyl esters to give quinazolin-4(3H)-ones in a one-pot synthesis.
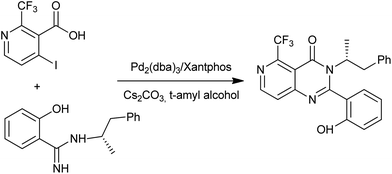 |
| | Scheme 28 Synthesis of pyrido[4,3-d]pyrimidin-4(3H)-one. | |
2.9 Palladium-catalyzed carbonylative synthesis of 4(3H)-quinazolinones
Since the original works from Heck and co-workers in 1974, Pd-catalyzed carbonylations have experienced impressive progresses during the last decades.63 In nowadays, Pd-catalyzed carbonylative transformation has already become a unique, powerful, and versatile tool for the synthesis of carbonyl containing heterocyclic compounds. In this contribution, Alper's group has developed an effective Pd-catalyzed three-component reaction of o-iodoanilines, imidoyl chlorides, and carbon monoxide affording substituted quinazolin-4(3H)-ones bearing a variety of functional groups in 2008 (Scheme 29).64 The same group also demonstrated a simple and efficient strategy for the one-step synthesis of potentially important 2-heteroquinazolin-4(3H)-one derivatives by palladium-catalyzed intermolecular addition and intramolecular cyclocarbonylation cascade reaction protocol.65 Based on the same strategy, an elegant domino process has also been developed for the synthesis of quinazolino[3,2-a]quinazolinones by forming five new bonds in a single step.66
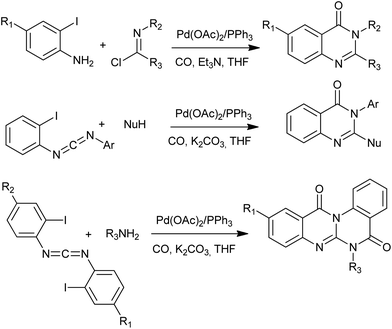 |
| | Scheme 29 Alper's work on Pd-catalyzed carbonylative synthesis of 4(3H)-quinazolinones. | |
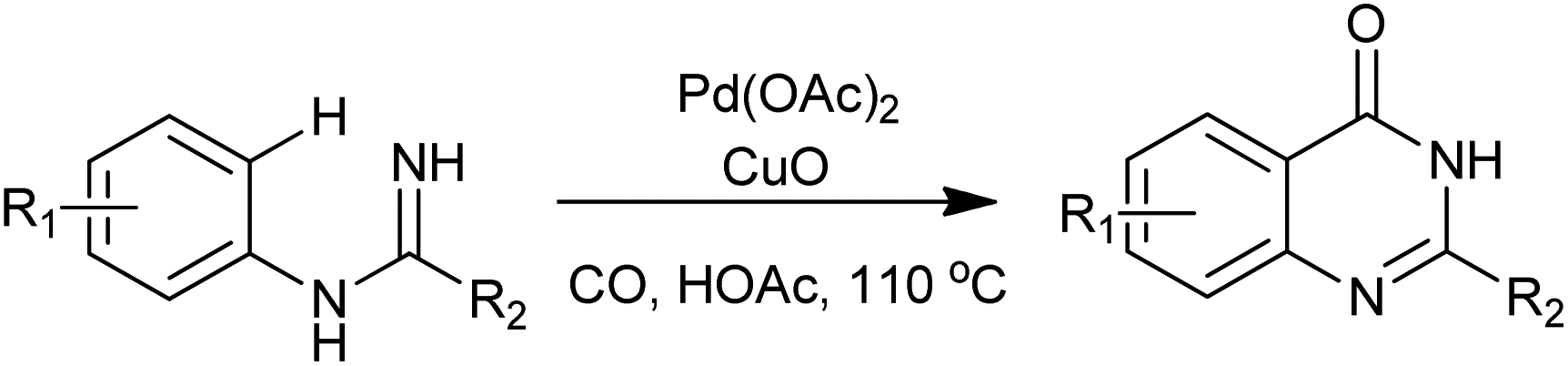
An interesting Pd-catalyzed intramolecular C(sp2)–H carboxamidation was reported by Zhu and co-workers (Scheme 30). The reactions were carried out in acetic acid, under one bar of CO together with one equivalent of CuO, various quinazolinones were produced from N-arylamidines in good yields.67 Compared with existing approaches to quinazolin-4(3H)-ones, this strategy features atom-economy and step-efficiency as no atoms except protons in substrates are lost during the process.
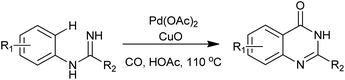 |
| | Scheme 30 Synthesis of 4(3H)-quinazolinones via Pd(II)-catalyzed C–H carboxamidation of N-arylamidines. | |
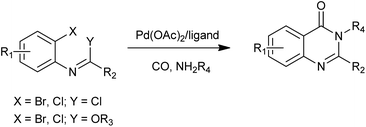
Recently, Willis et al. demonstrated that N-(o-halophenyl)imidoyl chlorides or imidates can be utilized as complementary precursors for the synthesis of quinazolinones by incorporation of a palladium-catalyzed aminocarbonylation reaction workers (Scheme 31).68 Under atmospheric pressure of CO, 2,3-disubstituted quinazolinones were produced in good to excellent yields. The use of amine nucleophiles bearing a range of substituents delivered quinazolinones in high yields.
 |
| | Scheme 31 Synthesis of 2,3-disubstituted quinazolinones from N-(o-halophenyl)imidoyl chlorides or imidates. | |
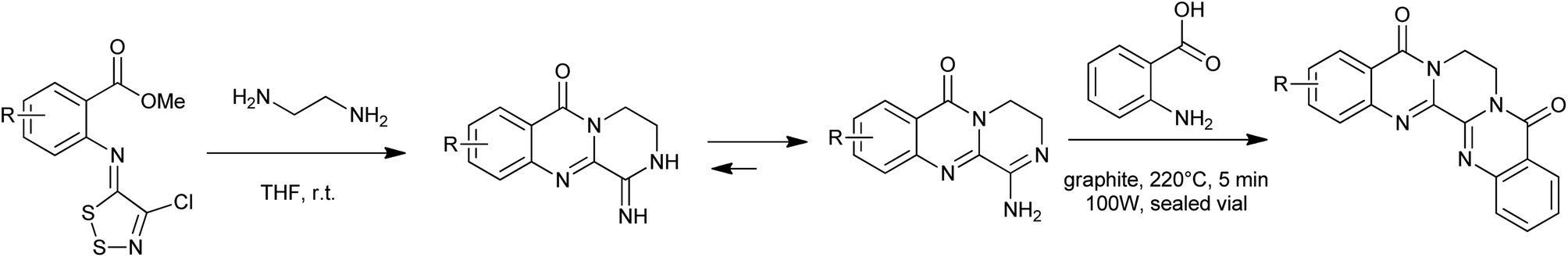
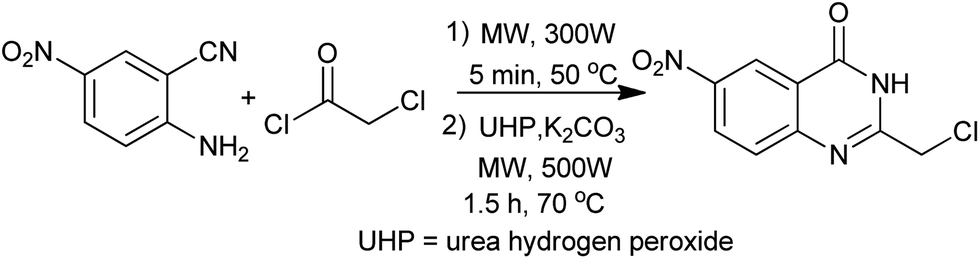
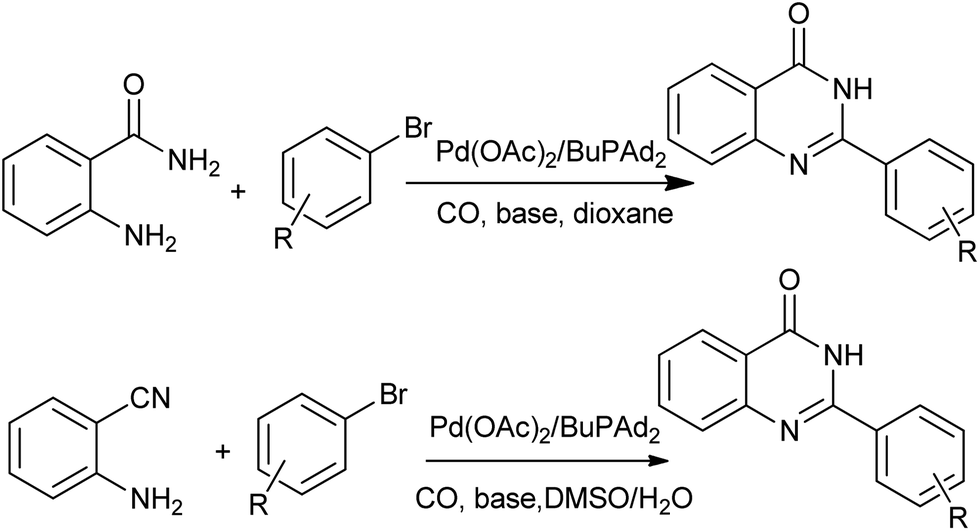
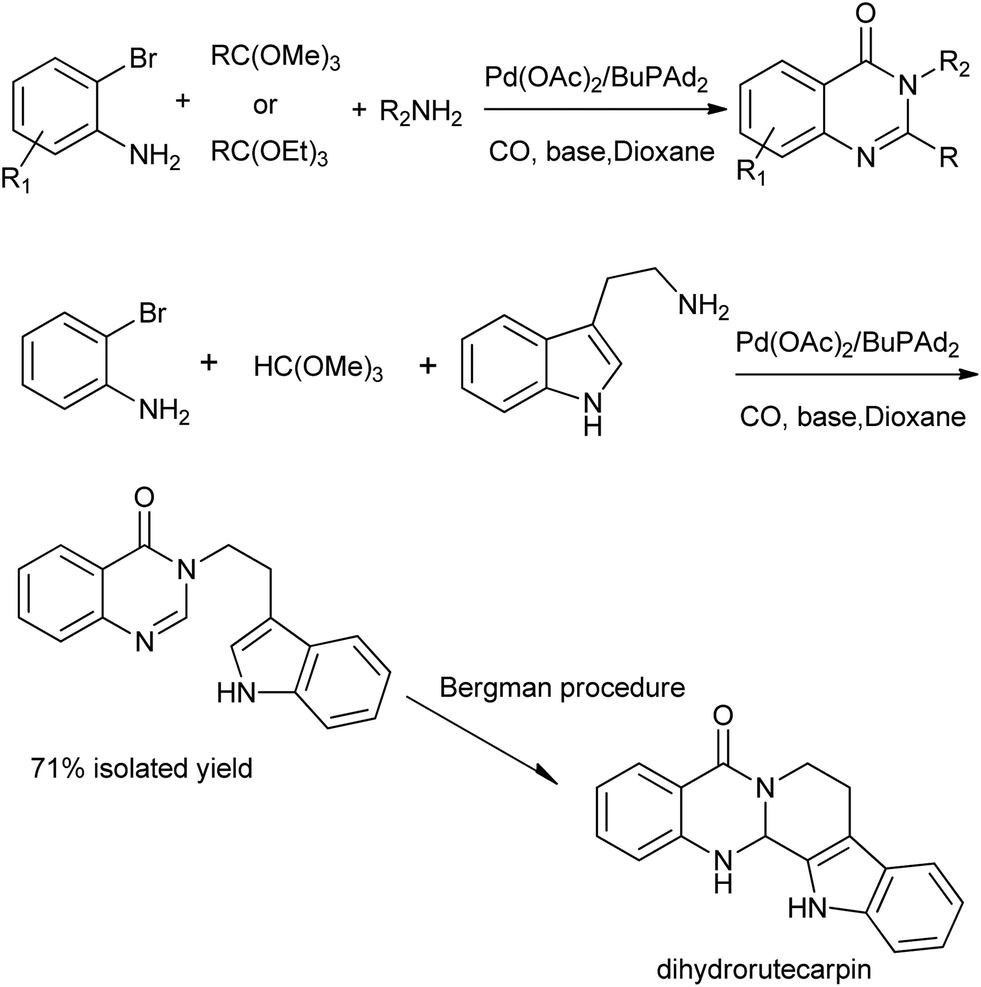
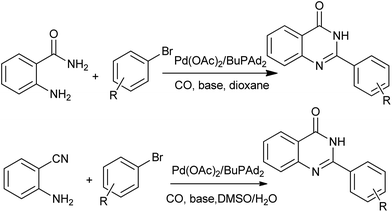
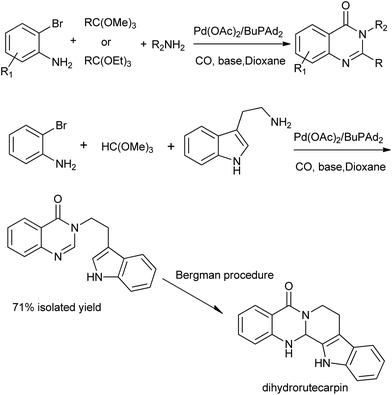
A straightforward procedure for the carbonylative synthesis of quinazolinones from aryl bromides and 2-aminobenzamide has been developed by our group (Scheme 32).69 Significant variation of the substrates allowed for the preparation of structurally diverse quinazolinone products. We envisaged that, as an appealing alternative to 2-aminobenzamide, 2-aminobenzonitrile can be applied as substrates in quinazolinones synthesis via in situ hydration of the nitrile group into amide. Followed by the guidance of this idea, we succeeded in preparation of quinazolinones from 2-aminobenzonitriles and aryl bromides using K2CO3 as an inexpensive base in aqueous solution.70 Given the importance of quinazolinones and carbonylative transformations, we recently developed a palladium-catalyzed four-component carbonylative coupling system for the synthesis of 4(3H)-quinazolinone in a concise and convergent fashion (Scheme 33).71 Starting from commercially available 2-bromoanilines, amines and ortho-esters and CO, the desired products were isolated in good yields in the presence of Pd(OAc)2/BuPAd2. Of significant practical importance is that the process tolerates a wide variety of synthetically useful functional groups, thus opening up a new avenue for efficient and sustainable production of highly functionalized 4(3H)-quinazolinone compounds. The method was used in the successful synthesis of the precursor for the bioactive dihydrorutaempine (Scheme 33).
 |
| | Scheme 32 Three-component Pd-catalyzed carbonylative synthesis of 4(3H)-quinazolinones. | |
 |
| | Scheme 33 Four-component Pd-catalyzed carbonylative synthesis of 4(3H)-quinazolinones. | |
3. Conclusion
In the present review, we attempted to highlight the recent development in the synthesis of 4(3H)-quinazolinone, which is a privileged scaffold in the pharmaceutical community for their therapeutic potential in treating a number of diseases. The complementarity of concise approaches facilitates the generation of libraries of functionalized 4(3H)-quinazolinone derivatives.
Acknowledgements
The authors thank the state of Mecklenburg-Vorpommern and the Bundesministerium für Bildung und Forschung (BMBF) for financial support. The general supports from Prof. Matthias Beller are also appreciated.
Notes and references
- S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS PubMed.
- P. Griess, J. Prakt. Chem., 1869, 369–371 Search PubMed.
- J. B. Koepfli, J. A. Brockman Jr and J. Moffat, J. Am. Chem. Soc., 1950, 72, 3323 CrossRef CAS.
- I. K. Kacker and S. H. Zaheer, J. Indian Chem. Soc., 1951, 28, 344–346 CAS.
- T. Panneer Selvam and P. V. Kumar, Res. Pharm., 2011, 1, 1–21 CAS.
- J. P. Michael, Nat. Prod. Rep., 2004, 21, 650–668 RSC.
- R. Arora, A. Kapoor, N. S. Gill and A. C. Rana, Int. Res. J. Pharm., 2011, 2, 22–28 CAS.
- A. Chawla and C. Batra, Int. Res. J. Pharm., 2013, 4, 49–58 CrossRef PubMed.
- D. J. Connolly, D. Cusack, T. P. O'Sullivan and P. J. Guiry, Tetrahedron, 2005, 61, 10153–10202 CrossRef CAS PubMed.
- S. Eguchi, Top. Heterocycl. Chem., 2006, 6, 113–156 CAS.
- F.-R. Alexandre, A. Berecibar and T. Besson, Tetrahedron Lett., 2002, 43, 3911–3913 CrossRef CAS.
- M. D. F. Pereira, V. Thiery and T. Besson, Tetrahedron, 2006, 63, 847–854 CrossRef PubMed.
- M. D. F. Pereira, L. Picot, J. Guillon, J.-M. Leger, C. Jarry, V. Thiery and T. Besson, Tetrahedron Lett., 2005, 46, 3445–3447 CrossRef CAS PubMed.
- K. R. Desai and A. R. Desai, Chem.: Indian J., 2005, 2, 216–220 CAS.
- Y. Kabri, A. Gellis and P. Vanelle, Green Chem., 2009, 11, 201–208 RSC.
- M. K. Kathiravan, R. R. Jalnapurkar, A. S. Chothe, T. S. Chitre, R. S. Tamboli and K. V. Srinivasan, Green Sustainable Chem., 2011, 1, 12–18 CrossRef CAS.
- T. Sugimori, T. Okawa, S. Eguchi, E. Yashima and Y. Okamoto, Chem. Lett., 1997, 869–870 CrossRef CAS.
- J. A. Bleda, P. M. Fresneda, R. Orenes and P. Molina, Eur. J. Org. Chem., 2009, 2009, 2490–2504 CrossRef.
- X.-H. Yang, M.-H. Wu, S.-F. Sun, M.-W. Ding, J.-L. Xie and Q.-H. Xia, J. Heterocycl. Chem., 2008, 45, 1365–1369 CrossRef CAS.
- M. M. Vögtle and A. L. Marzinzik, QSAR Comb. Sci., 2004, 23, 440–459 Search PubMed.
- C. Xie, H. X. Li, M. G. Liu and M. W. Ding, Chin. Chem. Lett., 2008, 19, 505–508 CrossRef CAS PubMed.
- W. M. Welch, F. E. Ewing, J. Huang, F. S. Menniti, M. J. Pagnozzi, K. Kelly, P. A. Seymour, V. Guanowsky, S. Guhan, M. R. Guinn, D. Critchett, J. Lazzaro, A. H. Ganong, K. M. DeVries, T. L. Staigers and B. L. Chenard, Bioorg. Med. Chem. Lett., 2001, 11, 177–181 CrossRef CAS.
- A. Kamal, E. V. Bharathi, M. J. Ramaiah, D. Dastagiri, J. S. Reddy, A. Viswanath, F. Sultana, S. N. C. V. L. Pushpavalli, M. Pal-Bhadra, H. K. Srivastava, G. N. Sastry, A. Juvekar, S. Sen and S. Zingde, Bioorg. Med. Chem., 2010, 18, 526–542 CrossRef CAS PubMed.
- J.-F. Liu, J. Lee, A. M. Dalton, G. Bi, L. Yu, C. M. Baldino, E. McElory and M. Brown, Tetrahedron Lett., 2005, 46, 1241–1244 CrossRef CAS PubMed.
- J.-F. Liu, P. Ye, B. Zhang, G. Bi, K. Sargent, L. Yu, D. Yohannes and C. M. Baldino, J. Org. Chem., 2005, 70, 6339–6345 CrossRef CAS PubMed.
- T. Güngör, Y. Chen, R. Golla, Z. Ma, J. R. Corte, J. P. Northrop, B. Bin, J. K. Dickson, T. Stouch, R. Zhou, S. E. Johnson, R. Seethala and J. H. M. Feyen, J. Med. Chem., 2006, 49, 2440–2455 CrossRef PubMed.
- M. R. Yadav, S. T. Shirude, A. Parmar, R. Balaraman and R. Giridhar, Chem. Heterocycl. Compd. (N. Y., NY, U. S.), 2006, 42, 1038–1045 CrossRef CAS PubMed.
- S. Boyapati, U. Kulandaivelu, S. Sangu and M. R. Vanga, Arch. Pharm. (Weinheim, Ger.), 2010, 343, 570–576 CrossRef CAS PubMed.
- D. Gupta, R. Kumar, R. K. Roy, A. Sharma, I. Ali and M. Shamsuzzaman, Med. Chem. Res., 2013, 22, 3282–3288 CrossRef CAS.
- V. Alagarsamy and G. Saravanan, Med. Chem. Res., 2013, 22, 1711–1722 CrossRef CAS.
- A. R. Khosropour, I. Mohammadpoor-Baltork and H. Ghorbankhani, Tetrahedron Lett., 2006, 47, 3561–3564 CrossRef CAS PubMed.
- K. Ighilahriz, B. Boutemeur, F. Chami, C. Rabia, M. Hamdi and S. M. Hamdi, Molecules, 2008, 13, 779–789 CrossRef CAS.
- M. M. Heravi, S. Sadjadi, N. Mokhtari Haj, H. A. Oskooie, R. H. Shoar and F. F. Bamoharram, Tetrahedron Lett., 2009, 50, 943–945 CrossRef CAS PubMed.
- M. M. Heravi, S. Sadjadi, S. Sadjadi, H. A. Oskooie and F. F. Bamoharram, Ultrason. Sonochem., 2009, 16, 708–710 CrossRef CAS PubMed.
- M. Wang, Z. G. Song and T. T. Zhang, Chin. Chem. Lett., 2010, 21, 1167–1170 CrossRef CAS PubMed.
- B. Wang, Z. Li, X. Wang, J. Tan, L. Gu and Z. Huang, Chin. Chem. Lett., 2011, 22, 951–953 CrossRef CAS PubMed.
- N. Saemian, O. K. Arjomandi and G. Shirvani, J. Labelled Compd. Radiopharm., 2009, 52, 453–456 CrossRef CAS.
- M. Bakavoli, O. Sabzevari and M. Rahimizadeh, Chin. Chem. Lett., 2007, 18, 1466–1468 CrossRef CAS PubMed.
- M. Bakavoli, A. Shiri, Z. Ebrahimpour and M. Rahimizadeh, Chin. Chem. Lett., 2008, 19, 1403–1406 CrossRef CAS PubMed.
- C. Zhang, C. K. De, R. Mal and D. Seidel, J. Am. Chem. Soc., 2008, 130, 416–417 CrossRef CAS PubMed.
- M. T. Richers, C. Zhao and D. Seidel, Beilstein J. Org. Chem., 2013, 9, 1194–1201 CrossRef CAS PubMed.
- H. Hioki, K. Matsushita, S. Nakamura, H. Horiuchi, M. Kubo, K. Harada and Y. Fukuyama, J. Comb. Chem., 2008, 10, 620–623 CrossRef CAS PubMed.
- N. Mulakayala, B. Kandagatla, Ismail, R. K. Rapolu, P. Rao, C. Mulakayala, C. S. Kumar, J. Iqbal and S. Oruganti, Bioorg. Med. Chem. Lett., 2012, 22, 5063–5066 CrossRef CAS PubMed.
- M. Sharif, J. Opalach, P. Langer, M. Beller and X.-F. Wu, RSC Adv., 2014, 4, 8–17 RSC.
- J. Zhou and J. Fang, J. Org. Chem., 2011, 76, 7730–7736 CrossRef CAS PubMed.
- J. Fang and J. Zhou, Org. Biomol. Chem., 2012, 10, 2389–2391 CAS.
- H. Hikawa, Y. Ino, H. Suzuki and Y. Yokoyama, J. Org. Chem., 2012, 77, 7046–7051 CrossRef CAS PubMed.
- H. Wang, X. Cao, F. Xiao, S. Liu and G.-J. Deng, Org. Lett., 2013, 15, 4900–4903 CrossRef CAS PubMed.
- A. Servais, M. Azzouz, D. Lopes, C. Courillon and M. Malacria, Angew. Chem., Int. Ed., 2007, 46, 576–579 CrossRef CAS PubMed.
- M.-H. Larraufie, C. Courillon, C. Ollivier, E. Lacote, M. Malacria and L. Fensterbank, J. Am. Chem. Soc., 2010, 132, 4381–4387 CrossRef CAS PubMed.
- A. Beaume, C. Courillon, E. Derat and M. Malacria, Chem.–Eur. J., 2008, 14, 1238–1252 CrossRef CAS PubMed.
- Y.-F. Wang, F.-L. Zhang and S. Chiba, Org. Lett., 2013, 15, 2842–2845 CrossRef CAS PubMed.
- F.-L. Zhang, Y.-F. Wang and S. Chiba, Org. Biomol. Chem., 2013, 11, 6003–6007 CAS.
- X. Liu, H. Fu, Y. Jiang and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 348–351 CrossRef CAS PubMed.
- D. Yang, H. Fu, L. Hu, Y. Jiang and Y. Zhao, J. Comb. Chem., 2009, 11, 653–657 CrossRef CAS PubMed.
- W. Xu, Y. Jin, H. Liu, Y. Jiang and H. Fu, Org. Lett., 2011, 13, 1274–1277 CrossRef CAS PubMed.
- T. Liu, C. Zhu, H. Yang and H. Fu, Adv. Synth. Catal., 2012, 354, 1579–1584 CrossRef CAS.
- X. D. Zhang, D. J. Ye, H. F. Sun, D. L. Guo, J. Wang, H. Huang, X. Zhang, H. L. Jiang and H. Liu, Green Chem., 2009, 11, 1881–1888 RSC.
- D. Guo, H. Huang, Y. Zhou, J. Xu, H. Jiang, K. Chen and H. Liu, Green Chem., 2010, 12, 276–281 RSC.
- L. Yu, M. Wang, P. Li and L. Wang, Appl. Organomet. Chem., 2012, 26, 576–582 CrossRef CAS.
- L. Xu, Y. Jiang and D. Ma, Org. Lett., 2012, 14, 1150–1153 CrossRef CAS PubMed.
- B. Li, L. Samp, J. Sagal, C. M. Hayward, C. Yang and Z. Zhang, J. Org. Chem., 2013, 78, 1273–1277 CrossRef CAS PubMed.
- A. Schoenberg and R. F. Heck, J. Am. Chem. Soc., 1974, 96, 7761–7764 CrossRef CAS.
- Z. Zheng and H. Alper, Org. Lett., 2008, 10, 829–832 CrossRef CAS PubMed.
- F. Zeng and H. Alper, Org. Lett., 2010, 12, 1188–1191 CrossRef CAS PubMed.
- F. Zeng and H. Alper, Org. Lett., 2010, 12, 3642–3644 CrossRef CAS PubMed.
- B. Ma, Y. Wang, J. Peng and Q. Zhu, J. Org. Chem., 2011, 76, 6362–6366 CrossRef CAS PubMed.
- J. E. Sadig, R. Foster, F. Wakenhut and M. C. Willis, J. Org. Chem., 2012, 77, 9473–9486 CrossRef CAS PubMed.
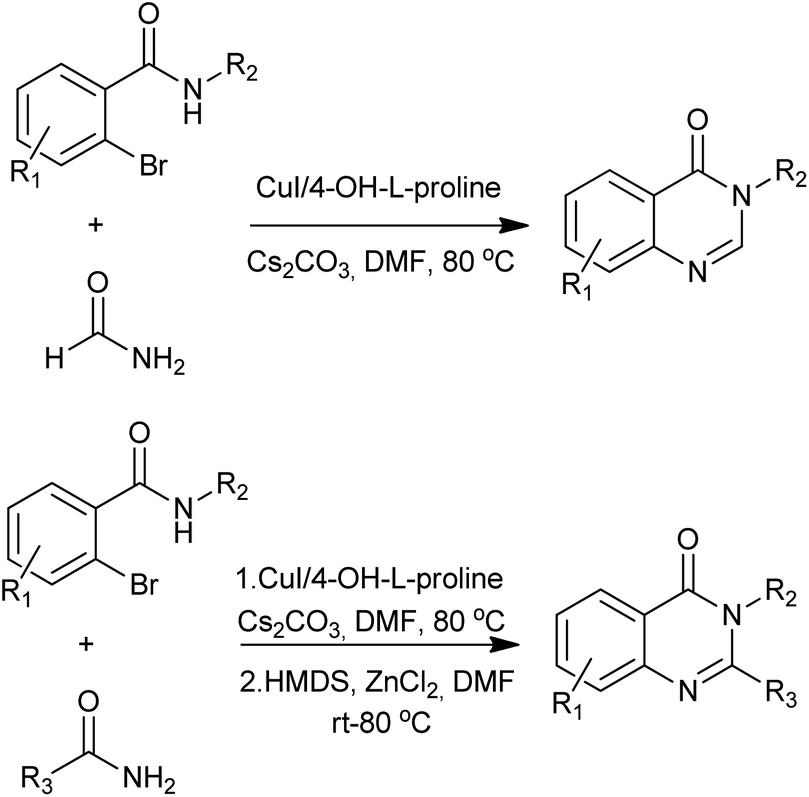
- X.-F. Wu, L. He, H. Neumann and M. Beller, Chem.–Eur. J., 2013, 19, 12635–12638 CrossRef CAS PubMed.
- H. Li, L. He, H. Neumann, M. Beller and X.-F. Wu, Green Chem., 2014 10.1039/c1033gc42089b.
- L. He, H. Li, H. Neumann, M. Beller and X.-F. Wu, Angew. Chem., Int. Ed., 2014, 53, 1420–1424 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.